Las talasemias son anemias hereditarias causadas por la falla en la síntesis de la hemoglobina en las cadenas alfa o beta de la globina.
La hemoglobina del adulto está normalmente compuesta por 98% de Hgb A1 que consta de dos cadenas alfa y dos cadenas beta; la alfa se localiza en el cromosoma 16 y el gen de la beta globina se localiza en el cromosoma 11. La Hgb A2 es el 1-2% de la hemoglobina normal y tiene dos cadenas alfa y dos delta. La Hgb F es menos de 1 % y esta formada por dos cadenas alfa y dos cadenas gamma.
La alfa talasemia es debida a la alteración en la síntesis de alfa globina. La severidad depende de la alteración en genes: la pérdida de una cadena alfa-gen produce un leve cambio microcítico en las células rojas y anemia mínima. La pérdida de dos cadenas alfa produce anemia moderada con microcitosis; puede haber esplenomegalia moderada pero no altera el estado funcional físico y la capacidad laboral es normal. La pérdida de tres cadenas alfa ocasiona severa anemia y la necesidad de transfusiones, hay esplenomegalia importante.
(Lea También: Síndrome Anémico, Anemia Macrocítica)
La falta de las cuatro cadenas alfa es incompatible con la vida.
Síndromes alfa talasémicos |
|||
Cadena |
Síndrome |
Htco |
VCM |
| 4 3 2 1 0 |
Normal Portador Talasemia menor Hgb fetal Hidrops fetalis |
Normal Normal 32-40% 22-32 |
Normal Normal 60-75 60-70 |
La hemoglobina H muestra alta afinidad por el O2 y produce pobre oxigenación de los tejidos.
La pérdida de las cuatro cadenas produce deprivación de oxígeno severa que lleva a falla cardiaca (hidrops fetalis) y muerte.
Estas formas de talasemia ocurren en Asia y Tailandia; es de alta prevalencia, y afecta a numerosos individuos llevando a problemas de salud pública en esas regiones. La pérdida de una cadena es probablemente la más frecuente y afecta aproximadamente 30% de personas de origen africano.
En las beta talasemias hay mutaciones que reducen la producción de cadenas beta con múltiples mutaciones. Clínicamente se describen como Beta-Talasemia menor, intermedia y mayor, dependiendo de la severidad de la anemia. La talasemia mayor afecta entre 30.000 a 60.000 personas recién nacidas de cada año; con frecuencia afecta individuos con ancestro italiano y griego.
La presentación común es en jóvenes con microcitosis, anemia, y esplenomegalia de tamaño variado de acuerdo a la severidad y requieren transfusiones frecuentes. En microcitosis la primera sospecha es ferropenia pero se debe considerar talasemia si no responde a suplencia o si no hay evidencia de pérdida de sangre.
En beta talasemia menor el paciente tiene una anemia modesta, microcitosis y recuento de glóbulos rojos alto o normal. Un elevado nivel de A2 establece diagnóstico de beta talasemia.
Si A2 es normal el diagnóstico más seguro es alfa talasemia. El análisis de ADN es la prueba clave y raramente es necesario para el diagnóstico.
La hemoglobina E es frecuente en individuos de la India o sudeste asiático; se diagnostica con una electroforesis de hemoglobina. Este desorden es similar a las talasemias menores y no requiere terapia.
El diagnóstico usando análisis de ADN es posible para alfa y beta talasemias. Un estudio cromosómico debe hacerse para consejería genética y constituye la base para terapia preventiva.
La sobredosis de hierro puede conducir a efectos nocivos con falla endocrina, cirrosis hepática, falla cardiaca y muerte.
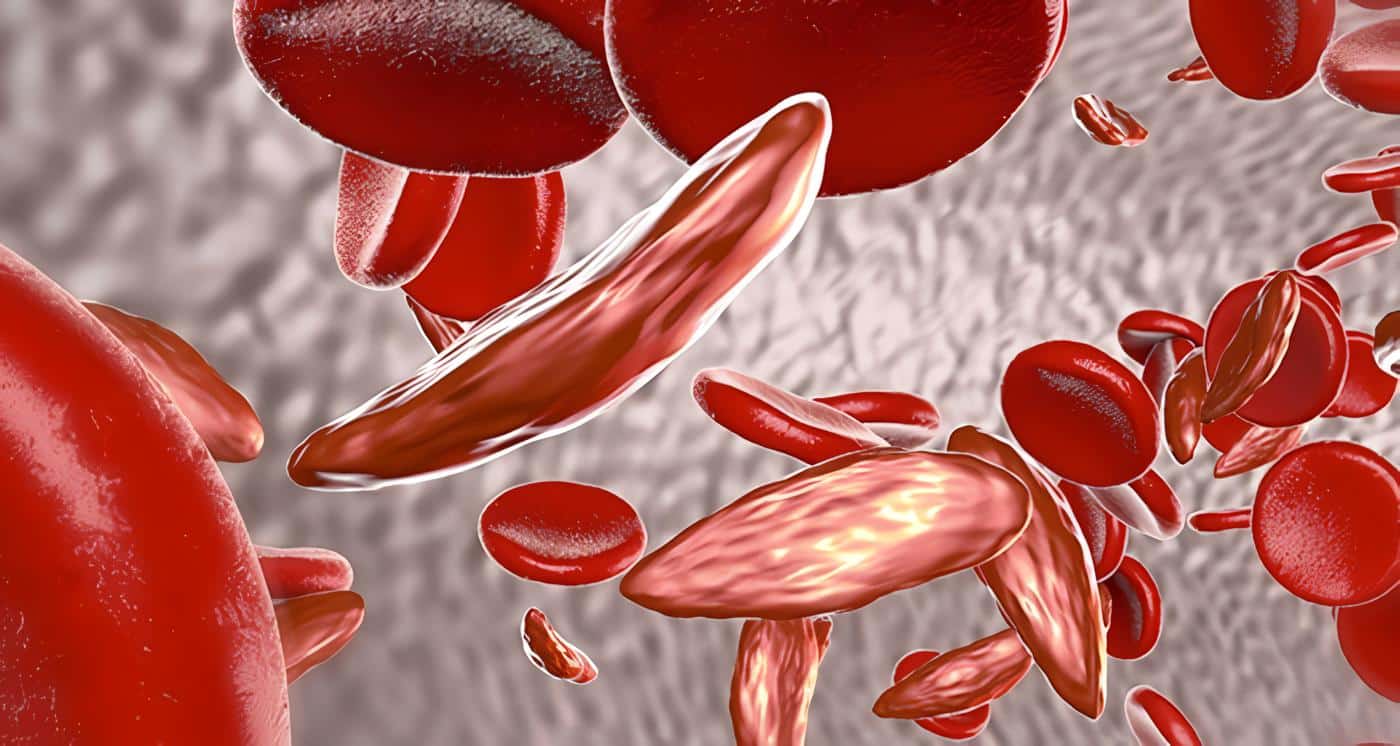
Anemia de células falciformes
La anemia de células falciformes es un desorden autosómico recesivo con una hemoglobina anormal que produce hemólisis crónica.
Es producido por una mutación puntual. Una simple cadena de ADN cambia un aminoácido y sustituye una valina por glutamina en la sexta posición en la cadena beta dando lugar a un tretamero denominado HgbS que, expuesta a bajas presiones de oxígeno, produce alteración en la membrana del glóbulo rojo llevando a cambios falciformes y hemólisis.
Hallazgos clínicos
La anemia hemolítica crónica produce ictericia, cálculos biliares, esplenomegalia y úlceras dolorosas en miembros inferiores. Puede poner en peligro la vida del paciente en las crisis hemolíticas severas o crisis aplásicas cuando la capacidad compensatoria de la médula está alterada por infección o deficiencia de ácido fólico.
Puede haber episodios de dolor agudo debido a oclusión de los vasos por hemólisis espontánea o inducida por deshidratación, infección o hipoxia. Estos episodios de vaso-oclusión pueden durar horas o varios días y se acompañan de fiebre baja. Los sitios mas afectados por dolor son huesos largos y tórax; cuando la oclusión es aguda y afecta los vasos sinusales pueden producir priapismo.
Cuando los episodios de hemólisis y vasooclusión son repetidos puede haber susceptibilidad a osteomielitis secundaria a estafilococo y menos común a Salmonella. Aparece hematuria por infartos en medula renal y puede haber retinopatía similar a la de diabetes.
Al examen físico el paciente a menudo se encuentra ictérico; hay hepatomegalia pero el bazo no es palpable en el adulto. El corazón está grande y se oye soplo sistólico con hiperdinamia.
Puede haber retinopatía y ulceras en miembros inferiores.
La expectativa de vida del paciente está disminuida y se considera la anemia de células falciformes como una enfermedad sistémica crónica.
El hematocrito se encuentra entre 20-30% y hay hemólisis crónica. En frotis de sangre periférica se observa células nucleadas, reticulocitosis de 10-25%, y presencia de células falciformes que compromete de 5 a 50% de glóbulos rojos.
Hay bilirrubina elevada, leucocitosis entre 12 a 15.000x mm3 y trombocitosis (Ver cuadro).
En pacientes heterocigotos con genotipo AS hay rasgo falciforme, pueden ser clínicamente normales y presentan episodios de dolor solo en condiciones extremas tales como ejercicio fuerte y elevadas alturas o viaje en cabina no presurizada. No se encuentra anemia y las células rojas son normales en el frotis periférico. El test de falciformia está positivo y electroforesis de hemoglobina muestra Hgb S en 40%.
Hemoglobinas / síndromes de cel falciforme |
|||||
Genotipo |
Dx Clínico |
H % |
AH % |
SH% |
A2H F% |
| AA AS SS SB* Talasem SB+Talasem As Atalasem |
Normal Rasgo falciforme Anemia falciforme Btalasem-falciforme Btalasem-falciforme Rasgo falc |
97-99 60 0 0 10-20 70-75 |
0 40 86-98 70-80 60-75 25-30 |
1-2 1-2 1-3 3-5 3-5 1-2 |
<1 <1 5-15 10-20 10-20 <1 |
Pacientes con anemia falciforme homocigotos y alfa talasemia tienen a menudo formas moderadas de hemólisis. Pacientes con doble presentación heterocigota para falciformia y beta talasemia están clínicamente afectados por hemólisis. Pueden presentar veno-oclusión por crisis que a menudo no son severas.
Tratamiento
No hay tratamiento específico, se recomienda suplencia de ácido fólico y la vacunación para neumococo que reduce la incidencia de infección.
En crisis hemolítica se recomienda transfusión.
Se debe mantener hidratación adecuada y se debe dar oxigeno en pacientes con hipoxia.
La exanguino-transfusión se recomienda en caso de crisis con veno-oclusión aguda, especialmente si hay crisis dolorosa intratable, priapismo y eventos cerebrales.
En caso de crisis frecuentes se recomienda uso de hidroxiurea 500-700 mg/día reduce el dolor y la frecuencia. El trasplante alogénico de médula ósea se constituye en opción para pacientes muy afectados.
La consejería genética es recomendable y es una estrategia razonable en prevención de estos síndromes.