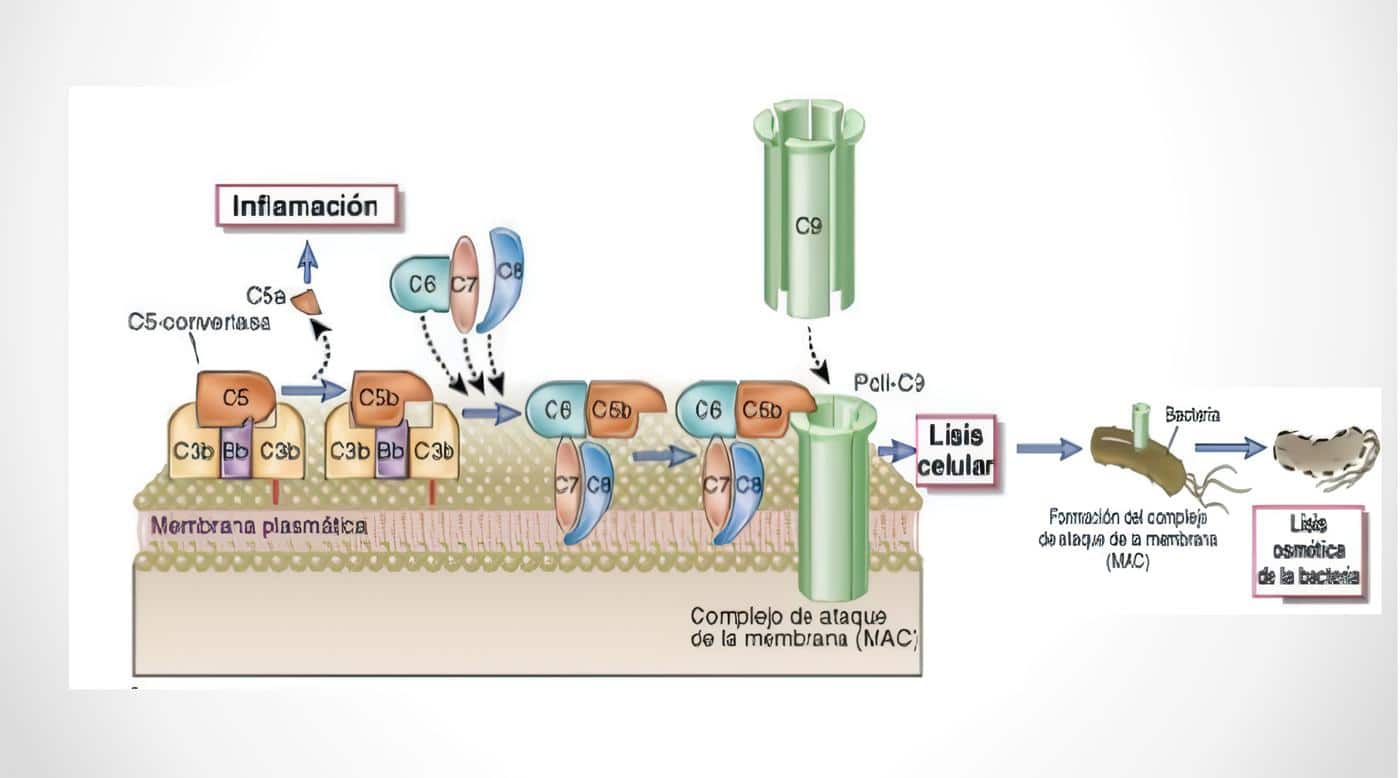
Complejo de Ataque a Membrana
Helí Salgado, Carlos J. Montoya, Juan A. López, Pablo J. Patiño
Grupo de inmunodeficiencias primarias, Facultad de Medicina – Universidad de Antioquia.”
El sistema del complemento agrupa un número importante de proteínas plasmáticas y de membrana que desempeñan un papel muy importante en la respuesta inmune inespecífica temprana; luego de su activación, se generan péptidos con notable actividad proinflamatoria y opsonizante, además de la formación del complejo de ataque a membrana con acción lítica directa.
Hasta el momento se han descrito deficiencias para la mayoría de las proteínas del complemento, sin embargo, corresponden al menor porcentaje (alrededor del 5%) de las inmunodeficiencias primarias reportadas.
Esto puede deberse a dos factores: rutinariamente sólo se evalúan el C3 y C4 en los pacientes sospechosos de una deficiencia del complemento, o no representan una causa importante de enfermedad porque la mayoría de estas alteraciones podrían ser muy leves y poco sintomáticas.
En la actualidad no existe una terapia específica para las deficiencias primarias del complemento, pero si hay una serie de medidas profilácticas y de atención que permiten a los afectados gozar de una aceptable calidad de vida.
Criterios Clínicos de Entrada en el complejo de ataque a membrana
- Cuadros de infección recurrente severos causados por gérmenes capsulados extracelulares (especialmente cocos Gram positivos, Haemophilus influenzae tipo B y neisserias) sugieren una deficiencia del factor C3 o de las proteínas del complejo de ataque a membrana; sin embargo, también deben considerarse en este caso las deficiencias de las células fagocíticas y las de anticuerpos.
- En pacientes con autoinmunidad que tienen asociada una infección recurrente por gérmenes extracelulares se debe descartar una deficiencia de los componentes activadores de la vía clásica (C2 y C4).
- Los cuadros de sepsis severa por meningococo o gonococo deben alertar sobre la presencia de una alteración en el complejo de ataque a membrana.
- La edad de inicio de las infecciones es variable según el componente alterado; por lo general las manifestaciones infecciosas en la deficiencia del factor C3 se inician en los primeros meses de vida; en otras alteraciones menos severas se encuentran en la edad adulta joven.
- Otros signos clínicos de alerta para descartar una deficiencia del complemento son: infecciones bacterianas sistémicas de evolución rápida y fulminante; el angioedema en individuos con antecedentes familiares de esta entidad (deficiencia del inhibidor del C1 esterasa); la hemoglobinuria paroxística nocturna (deficiencia del CD59).
Estudios Básicos de Laboratorio
Estudios de la Primera Etapa
- Hemoleucograma con sedimentación y extendido de sangre periférica: no hay alteraciones específicas de las deficiencias del complemento; en las infecciones recurrentes severas por deficiencias en estas proteínas se puede encontrar una anemia con características de cronicidad.
- Estudios para determinar la etiología de las infecciones y su sensibilidad. Las infecciones en las deficiencias del complemento por lo general son debidas a bacterias extracelulares como los Streptococcus, Neisserias y Haemophilus influenzae.
- Electroforesis de proteínas séricas: se debe tener en cuenta que los factores del complemento más importantes están representados en la fracción beta.
- Estudios imagenológicos cuando sean necesarios (Rx de tórax, Tomografía Axial Computarizada, etc.).
- Si se confirma un diagnóstico de inmunodeficiencia primaria, se debe realizar el árbol genealógico familiar con el mayor número de generaciones posibles.
Estudios de la Segunda Etapa
- Dosificación sérica de C3 y C4 (Tabla 1).
- Determinación de la capacidad hemolítica del complemento en el suero del paciente (CH50).
- En los casos de edema angioneurótico recurrente severo o con antecedentes familiares se debe dosificar en suero la concentración del inhibidor de C1 estearasa (C1INH).
- En los casos de hemoglobinuria paroxística nocturna, se debe evaluar la expresión de CD59 en glóbulos rojos por citometría de flujo.
- Debido a las implicaciones que tiene una inmunodeficiencia primaria, todo diagnóstico debe estar respaldado por una segunda prueba de laboratorio confirmatoria.
Estudios de la Tercera Etapa
- Análisis cuantitativo (ELISA) y funcional de los diferentes componentes del complemento.
- Estudios genético-moleculares para la caracterización de mutaciones en las diferentes deficiencias.
Manejo Terapéutico
Medidas generales en el complejo de ataque a membrana
En las deficiencias del complemento por ausencia del C3 o defectos en el complejo de ataque a membrana los individuos afectados presentan una mayor susceptibilidad al desarrollo de infecciones más severas que las observadas en los individuos inmunocompetentes.
Por ello, además de la terapia antimicrobiana, es fundamental inducir en el paciente y su familia la adopción de hábitos personales y sociales que permitan prevenir el desarrollo de nuevas infecciones o el agravamiento de las presentes. Algunas de las medidas más importantes son:
- Los individuos afectados deben ser advertidos de evitar cualquier exposición innecesaria a fuentes de infección (visitas a hospitales, contactos domiciliarios con familiares enfermos, etc.).
- Dieta balanceada que cubra plenamente las necesidades en proteínas, vitaminas y oligoelementos.
- El paciente no debe fumar ni consumir drogas ilegales (alcohol, etc.); debe evitar a toda costa constituirse en un fumador pasivo en su medio ambiente familiar y social.
- El afectado debe portar una tarjeta con la identificación clara de su problema, los riesgos y medidas para una situación de urgencia.
Tratamiento médico
- No existe terapia específica para ninguna de las deficiencias congénitas del complemento; sin embargo, un cuidadoso manejo de los afectados puede mejorar la calidad de vida y supervivencia de los afectados.
- En las deficiencias de C3 y del complejo de ataque a membrana se deben iniciar prontamente antibióticos cuando existan los primeros signos de infección (fiebre, síntomas generales, etc.).
- Cuando existe una deficiencia del C3 y el paciente presenta una infección severa se puede considerar la aplicación de plasma fresco (20 mL/kilo de peso), teniendo en cuenta la vida media corta y los bajos niveles del C3 en este derivado sanguíneo.
- La aplicación de plasma también es útil en las crisis de edema angioneurótico hereditario.
- Se recomienda la aplicación temprana (al paciente y sus familiares cercanos) de vacunas contra Haemophilus influenzae tipo B, neumococo y meningococo; el uso de penicilinas profilácticas a largo término sólo se justifica cuando la enfermedad por el meningococo es endémica en la zona.
Seguimiento de los pacientes
- Los pacientes con diagnóstico de inmunodeficiencia primaria se programan para un control mensual por consulta externa, para evaluar su estado clínico y supervisar la evolución desde el punto de vista de laboratorio; estas citas serán también de utilidad para las labores de educación y prevención dirigidas al paciente y la familia.
- Otros análisis de laboratorio están indicados cuando sean necesarios para el estudio de alguna enfermedad o condición asociada: directos y cultivos de secreciones; coprológico, pruebas de función hepática y renal; química sanguínea; estudios imagenológicos, etc.
- Se recomienda realizar anualmente una evaluación de: C3, C4, CH50 y autoanticuerpos (ANAs, etc.).
Referencias Bibliográficas
- 1. García de OD, Patiño PJ, Salgado H, et al. Evaluación del paciente con inmunodeficiencia. Síndrome de infección recurrente patológica. Medicina y Laboratorio. 1997; 7: 545-575.
- 2. Holland MS, Gallin JI. Evaluation of the patient with suspected immunodeficiency. In: Mandell, Douglas, Bennett, eds. Principles and Practice of Infectious Diseases. New York: Churchill Livingstone. 1995:149-158.
- 3. Paul ME, Shearer WT. The child who has recurrent infection. Immunol Allergy Clin North Am. 1999; 12 (2):423-436.
- 4. World Health Organization Scientific Group. Primary Immunodeficiency Diseases. Clin Exp Immunol. 1995; 99 (suppl. 1):1-24.
- 5. Sullivan KE, Winkelstein JA. Genetically determined deficiencies of the Complement System. En: Primary immunodeficiency diseases: A molecular and genetic approach. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford. 1999. pp: 397-416.
- 6. Stiehm ER. Conventional therapy of primary immunodeficiency diseases. En: Primary immunodeficiency diseases. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford. 1999. pp: 448-458.