Dr. Jaime F. Pérez Niño.
Miembro de Número Sociedad Colombiana de Urología.
Dra. Adriana Lema Izquierdo.
Endocrinóloga Pediatra.
Clínica Infantil Colsubsidio.
Bogotá. Colombia.
Paciente producto de un segundo embarazo, padres jóvenes no consanguíneos, parto por cesárea iterativa, peso 3440g talla 51cms. Al examen físico RN sana, con genitales externos femeninos.
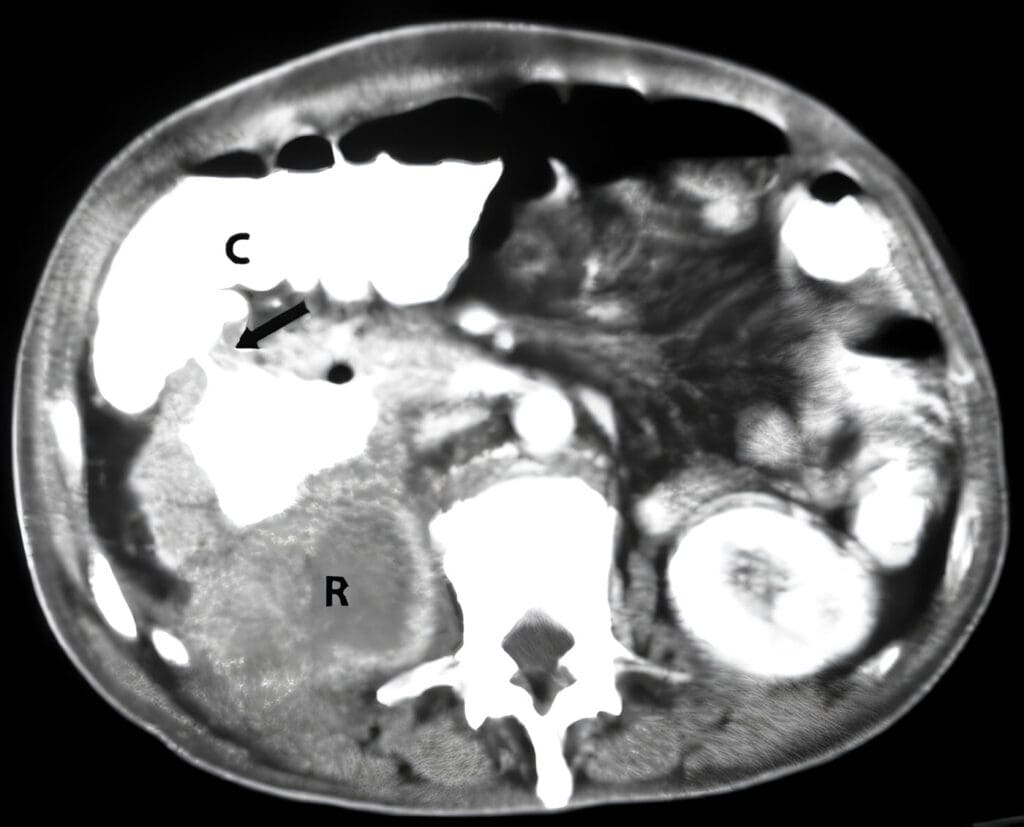
A los 20 días le diagnostican hernias inguinales bilaterales.En el acto operatorio se encontraron gónadas bilaterales de las cuales se tomaron biopsias. El reporte de patología mostró en los cortes tejido testicular con abundantes células de Leydig y túbulos pequeños con frecuentes espermatogonias y algunas células de Sertoli.
Se le realiza un cariotipo reportado como 46XY. Se hizo un diagnóstico de Testículo feminizante.
A los 4 meses se le practicó orquidectomía bilateral, con reporte de patología de testículos intraabdominales.
Se le diagnosticó además displasia bilateral de caderas tratada con férula de Milgram hasta los 2 años y medio, con buena respuesta al tratamiento.
Valoración por Endocrinología Pediátrica a los cuatro meses y medio; se encuentra una niña que mantiene un peso en P10-20 y talla en P55, velocidad de crecimiento normal, con genitales femeninos externos normales. Se hizo un diagnóstico de: Síndrome de Insensibilidad a los Andrógenos.
A los 4 años y medio se le practicó una genitografía y vaginoscopia que mostró una vagina con ausencia del tercio superior, de 4 cms de profundidad. En la actualidad no recibe ningún tratamiento médico y se iniciará el reemplazo hormonal en la época prepuberal.
El Síndrome de Insensibilidad a los Andrógenos (SIA) se describió en 1817 cuando en la autopsia de una mujer normal con amenorrea primaria se encontraron testículos intraabdominales. Posteriormente, Morris reportó 82 casos de SIA en 19531. La incidencia actual es de 1:20000 a 64:000 varones nacidos vivos, con una expresión fenotípica muy variable desde la forma completa (SIAC) hasta las parciales (SIAP) en hombres con fenotipo normal cuya única manifestación es la azoospermia2.
El diagnóstico puede ser muy evidente cuando tiene cariotipo 46XY con genitales externos femeninos; esto corresponde al 50-70% del diagnóstico del pseudohermafroditismo masculino, pero se deben descartar otras causas como disgenesia gonadal, hipoplasia de células de Leydig, defectos en la biosíntesis enzimática de andrógenos (déficit de 3 Beta hidroxiesteroide deshidrogenasa, déficit de 17 alfa hidroxilasa, déficit de 17 cetosteroide reductasa y de 5 alfa reductasa)3.
El receptor androgénico (RA) se localiza en el cromosoma X por lo cual el SIA está ligado a X4. El RA tiene 8 exones; las mutaciones pueden alterar la afinidad para ligar el andrógeno, la especificidad con otras hormonas esteroides, producir pérdida total de la función o requerir de concentraciones más altas de andrógenos. Pueden ser puntuales 82%, deleción completa 1% o parcial 4%3. Hasta el momento se han descrito 300 mutaciones2.En algunos casos no se detecta la mutación y es posible que otros factores intervengan en la activación transcripcional del RA5. No hay una correlación directa entre la mutación, el fenotipo y la alteración funcional.
La mutación del RA se ha asociado también a una atrofia muscular espino-bulbar ligada a X (Sindrome de Kennedy) que cursa con una severa degeneración axonal y de las fibras sensoriales, produciendo un patrón de neuropatía axonal distal central y periférica6.
Debido a que el SIA es un trastorno de carácter recesivo ligado a X, en el hombre un solo alelo produce un fenotipo alterado. Las mujeres portadoras tienen un fenotipo normal aunque se ha visto algun tipo de retraso puberal, disminución del vello púbico y axilar7.
Generalmente hay historia familiar de amenorrea o infertilidad, que se asocia en 64% con SIAC y 18% con SIAP.
Clínicamente, la hernia inguinal está presente en el 90% de las niñas con SIAC de las cuales el 31% es unilateral, 59% bilateral, mientras en el SIAP solo se encuentra en 12% de las pacientes. Se encuentran uno o dos testículos palpables en aproximadamente 80% de los caso con estudio histológico concordante8. Debido a la alta incidencia de hernia inguinal (1-12%) con el SIA las niñas con esta patología ameritan un cariotipo8.
En la pubertad la amenorrea primaria es la presentación más común, con masas labiales o inguinales presentes, genitales femeninos internos ausentes o diminutos, un tercio tiene remanentes de ducto mullerianos y ocasionalmente vestigios del Wolff3.
Los criterios diagnósticos son: hábito femenino con depósitos grasos femeninos normales, senos generalmente poco desarrollados, escaso vello púbico y axilar, genitales externos femeninos con labios menores poco desarrollados, clìtoris normal o pequeño y vagina normal, ausencia de genitales internos femeninos, gónadas con túbulos seminíferos sin espermatogénesis y estudio hormonal que sugiera que la presencia de testículos3.
El diagnóstico del SIAP es más complejo por su variedad fenotípica amplia. En la forma leve se ve un varón normal con infertilidad por oligospermia severa o azoospermia, la moderada se presenta en un varón mal virilizado con genitales pequeños, ginecomastia postpuberal y disminución de los caracteres sexuales secundarios (barba y vello corporal). Las formas severas presentan hipospadias y criptorquidia hasta el Síndrome de Reifenstein con genitales internos masculinos.
El diagnóstico prenatal con DNA trofoblástico se recomienda en pacientes con historia familiar. En la infancia cuando se sospecha el diagnóstico se debe realizar un cariotipo.
El estudio del RA por DNA recombinante puede mostrar los defector moleculares y el grado de disfunción que puede ser variable y no se correlaciona necesariamente con el fenotipo genital en la insensibilidad parcial. En pocos casos hay ausencia completa del receptor que se asocia con la insensibilidad completa.
El estudio hormonal varía con la edad. En los prepuberales con SIAC la LH y testosterona están en rangos normales; en los postpuberales con testículos in situ pueden tener una LH elevada con niveles normales o elevados de testosterona y estradiol.
En los casos de SIAP los niveles son normales, en algunos casos están elevados por falta de retroalimentación.
La hormona antimulleriana muestra la función de las células de Sertoli lo que podría ser útil como prueba de la existencia de tejido testicular9 y la medición de la SHBG (hormona ligante de esteroides sexuales) refleja la funcionalidad del Receptor Androgénico al administrar un esteroide anabólico para evaluar su generación in vivo10.
El tratamiento en el SIAC comprende la asignación femenina fenotípica, con la comprensión y aprobación de los padres; la orquidectomía que podría ser temprana, favorable por la corrección de la hernia y porque evita la malignización testicular (aunque no se ha visto en la prepubertad); o puberal, para favorecer los cambios del tejido mamario por la conversión de testosterona a estrógeno y prevenir la desmineralización ósea durante la infancia. La orquidectomía tardía tiene los inconvenientes de que la persistencia de la hernia o los testículos que puede ser incómoda o dolorosa y que enfrentar el diagnóstico en la pubertad, puede traer problemas psicológicos de adaptación y de identidad importantes.
Se debe iniciar reemplazo hormonal con dosis inicialmente bajas de estrógenos y posteriormente progestágenos, lo cual mejora la osteopenia descrita11,12.
El tratamiento del SIAP es más complejo ya que depende del fenotipo y de la respuesta de los tejidos al estímulo androgénico, según lo cual se hace la asignación.
En los varones postpuberales se debe manejar la infertilidad y vigilar los testículos ya que tienen una incidencia tumoral similar a la del testículo criptorquídico13.
Finalmente, el apoyo psicológico es indispensable para el paciente y su familia, con el fin de la aceptación de esta patología y lograr la mejor calidad de vida para estos pacientes.
Comentario
Con relación a nuestra paciente, no se le realizaron pruebas hormonales que hubieran servido para hacer diagnóstico diferencial con deficiencia de testosterona o de 5 alfa reductasa, porque el diagnóstico como en la gran mayoría de los casos de insensibilidad completa a los andrógenos se hizo intraoperatorio. De ahí la importancia de solicitar cariotipo preoperatorio en todas las niñas con hernia inguinal.
En los pacientes con antecedentes familiares en los que se hace diagnóstico prenatal de SIA, se debe plantear con un estricto criterio ético la justificación de terminar un embarazo, ya que la mutación no predice el fenotipo.
En el hombre adulto en estudio de infertilidad secundaria a SIA se debe plantear la disyuntiva de someterse a fertilización por medios de Inyección de Esperma Intracitoplasmática, ya que si posee una mutación ligada a X la transmite a todos sus hijos varones.
Finalmente, si existieran pruebas más sensibles para evaluar la respuesta clínica a la testosterona y dihidrotestosterona, se podría realizar la asignación sexual de una forma más apropiada y así evitar los problemas asociados a la reasignación de sexo.
Bibliografía
1. Morris. Syndrome of Testicular feminization in male pseudohermaphrodites. AM J Gynecol 1953, 65:1192.
2. Ahmed, S.F. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as Androgen Insensitivity Syndrome. JCEM 2000, 85:658-665.
3. Wiener JS. Molecular Biology and function of the androgen receptor in genital development. J of Urology 1997, 157:1337-86.
4. Brown C.J. Androgen receptor locus on the human X chromosome: regional localization to Xq11-12 and description of a DNA polymorphism. Am. J. Human Genet. 1989, 44:264.
5. Okabe T. Pathogenesis of androgen insensitivity syndrome. Nippon Rinsho 1998 Jul; 56(7):1881-6.
6. Andersen KV. X linked recessive bulbospinal neuronopathy-Kennedy´s Syndrome. Tidsskr Nor Laegeforen Apr, 1999, 119(11):1591-4
7. Quigley, C.A Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr. Rev. 1995,16:271.
8. Viner, RM. Androgen Insensitivity Syndrome: a survey of diagnostic procedures and management in the U.K. Arch Dis Child 1997(77)305-9.
9. Rey RA. Evaluation of Gonadal Function in 107 intersex patients by means of Serum Antimullerian Hormone Measurement. J Clin Endocrinol Metab 1999, 84:627-31.
10. Holterhus P-M. Mosaicism due to a Somatic Mutation of the androgen receptor gene determines phenotype in androgen insensitivity syndrome. J Clin Endocrinol Metab 1997, 82:3584-89.
11. Marcus R. The contribution of testosterone to skeletal development and maintenance: lessons from the androgen insensitivity syndrome. J Clin Endocrinol Metab 2000 Mar;85(3):1032-7.
12. Bertelloni S. Altered bone mineral density in patients with complete androgen insensitivity syndrome. Horm Res 1998,50(6):309-14.
13. Alvarez-Nava F. Complete androgen insensitivity syndrome: clinical and anatomopathological findings in 23 patients. Genet Couns 1997, 8(1):7-12.

