Resúmenes del XXXIV Congreso Nacional de Urología
Dr. Mauricio Coll
Dr. Marco A. Nossa
Dr. Efraim Bonilla
La diferenciación sexual se lleva a cabo en tres niveles determinados en la siguiente manera:
1. Sexo genético: Aparece desde el momento mismo de la fecundación. Se encuentra determinado por antígenos y genes codificadores de proteínas específicas.
* TDF: Brazo corto de cromosoma Y. Responsable del mecanismo inicial de la diferenciación testicular.
* Zona Fy y Fx: Proteínas que codifican para que la diferenciación sexual se lleve hacia uno u otro sexo.
* Sry: Descubierto en 1990. Se localiza en región eucromatérica distal del brazo corto del cromosoma Y. Es el gen más importante de la diferenciación sexual masculina.
Las mutaciones en Sry van a producir ambigüedad sexual en forma de disgenesia gonadal o reversión sexual completa.
Hay individuos que teniendo Sry, son fenotípicamente femeninos, hablándose entonces de un Locuz Z, que sería un regulador (-) de genes masculinos específicos.
* DSS: Gen importante en diferenciación sexual, ubicado en brazo corto de cromosoma X, responsable de la atribución de un sexo revertido en individuos XX.
* SF1: Se manifiesta tempranamente después de la fecundación, implantándose en gónadas, suprarrenal e hipotálamo. Alteraciones en SF1 determinan ausencia de gónadas y suprarrenal, con FSH y LH disminuidas.
* DAX-1: Ubicado en cromosoma X. Su alteración produce hipoplasia de suprarrenales y reversión sexual.
* TDA: Alteración a nivel de 9p24 en individuos XY, manifestando reversión sexual.
* XXT1: 11p13. Gen represor de T. Wilms. Se presenta como reversión sexual + T. Wilms, con diferentes niveles de severidad según síndrome (S. Derys-Drash, S. Frasser, S. Wagr).
2. Sexo gonádico: Después del sexo genético y hacia la 7 semana comienza la diferenciación gonadal, la cual es de forma activa en XY y pasiva en XX.
3. Sexo fenotípico: Una vez se ha diferenciado la gónada (testículo), se suceden cambios en estructuras genitales que han sido idénticas en ambos sexos hasta la 7 semana (C. Wolf, C. Muller, seno urogenital). En el hombre, por efecto de la testosterona, se desarrolla de forma activa el C. Wolf, produciéndose a la vez a nivel testicular, hormona antimulleriana que hace que se presente regresión de conducto de Muller. En ausencia de testosterona, habrá desarrollo en forma pasiva de C. Muller, e involución de C. Wolf.
Hiperplasia suprarrenal congénita
Deficiencia enzimática, (21 hidroxilasa, 11 hidroxilasa, 17 hidroxilasa) que se hereda de forma autosómica recesiva, y se traduce en un aumento en la concentración de andrógenos circulantes. Son pacientes genotípicamente femeninas, en su mayoría, que manifiestan alteraciones que van desde variaciones en el tamaño del tubérculo genital, hasta virilización completa. La hiperplasia adrenal congénita también se puede dar en individuos fenotípicamente masculino (pseudohermafroditismo masculino), por déficit de 21 hidroxilasa, caracterizándose en ellos, por trastornos hidroelectrolíticos (nefropatía perdedora de sal), pubertad precoz (macropene), y en algunos casos asociada a neoplasias, principalmente de hígado y glándula suprarrenal.
El estudio inicial de esta entidad debe incluir estudio familiar, examen físico exhaustivo (incluido TR), así como estudio hormonal (17, hidroxiprogesterona), cariotipo y genitografía. En la actualidad su diagnóstico se puede realizar «in vitro» (amniocentesis o técnicas de biología molecular), con la posibilidad de realizar manejos tempranos, con el objeto de disminuir los efectos de la enfermedad en el feto.
El tratamiento con hidrocortisona y antiandrógenos al momento de diagnóstico es fundamental, asociado al tratamiento quirúrgico temprano (clitoridoplastia reductora vs. genitolastia feminizante), dependiendo este último del nivel al que se encuentre el seno urogenital, y la edad del paciente al momento del diagnóstico.
Disgenesias gonadales y hermafroditismo
– Hermafrodita verdadero: Cariotipo: 46 XX(57%). 46XY (13%) Mosaico (30%).
Combinaciones posibles ovariotestículo (rara, ovotestis bilateral (+ fc), ovotestis-ovotestis, ovotestis-testículo.
– Testículo feminizante completo: Mujeres XX. Testosterona: normal, DHT, LH y estrógenos elevados.
Causas: falta de respuesta a la DHT (por carencia de receptores, o presencia de receptores pero ausencia de proteína transportadora de DHT). Se caracterizan por genitales externos femeninos, hipertrofia de clítoris, vagina de profundidad variable, testículos criptorquídicos (histológicamente casi normales), ausencia de estructuras mullerianas y mamas normales.
– Testículo feminizante incompleto: Mujer XY. Testosterona: N. DHT:
Causas: Deficiencia de 5 alfa reductasa (rara). Se caracteriza por presentar desarrollo genital femenino inicialmente, que hacia la pubertad comienza a presentar algún grado de masculinización.
- S. Reifenstein: Pene corto, atrofia testitular puberal, diferentes grados de hipospadias y ginecomastia.
- S. Lub: Genitales casi feminizados.
- S. Aiman: Genitales externos masculinos, esterilidad.
- S. de persistencia de Muller: Carioptipo: Mosaico: 45XY – 45X0.
Son en la mayoría de los casos niños, con pene bien desarrollado, pero ausencia de gónadas en escroto. Siempre se debe solicitar cistouretrografía durante la evolución inicial en busca de esbozos de vagina. Ante la ausencia de verum-montanum, se recomienda evaluación endoscópica.
– Disgenesia gonadal mixta: Siempre en Mosaico: 46XY – XY. Es la segunda causa de genitales ambiguos. Se requiere cromosoma Y para que se desarrolle testículo. Siempre tienen útero unicorne y vagina, así como testículo normal palpable unilateralmente en escroto y esbozo de gónada. 25% desarrollan gonadobastomas en primera década de la vida. El grado de hipospadias es variable. Evaluación: Cariotipo y genitografía.
– Disgenesia gonadal pura: Cariotipo: Mosaico 45X0/XO/XX-XY. Niñas con hipertrofia variable del clítoris (clitoromegalia), con esbozos gonadales bilaterales, que al momento de la pubertad no presentan menarquia, ausencia de bello púbico, desarrollo de genitales externos y glándula mamaria.
Discusión
El problema de niños con genitales ambiguos debe ser abordado de forma individual e interdisciplinaria (urólogo pediátra, cirujano pediátra, endocrinólogo pediátra, genetista, psicólogo y trabajador social). Hoy en día existe una legislación muy precisa al respecto, que adecuada o no, debe ser conocida por los profesionales que manejan este patología, ya que las implicaciones de tipo legal que pueden resultar de manejos no acordes con él pueden generar sanciones de tipo penal.
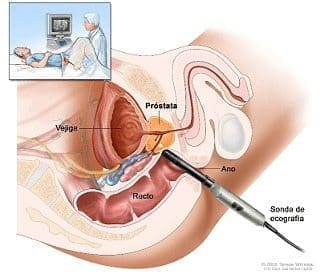
Prostatectomía perineal
Dr. Jaime Andrés Cajigas
Se presentó un video de una prostatectomía perineal paso a paso, invitando a la audiencia a tener en cuenta este abordaje quirúrgico, como una opción más en el tratamiento de los pacientes con cáncer de próstata localizado.
Indicaciones
• Pacientes que no sean candidatos a linfadenectomía pélvica.
• PSA Menor de 10 ng/ml.
• Gleason menor de 6.
• Menos de 3 biopsias prostáticas positivas.
Ventajas
• Acceso rápido.
• Anastomosis fácil.
• No tensión en la sutura de la anastomosis.
• Sangrado mínimo.
Desventajas
• Espacio reducido.
• Dificultad en la conservación de fascículos neurovasculares.
• Dificultad en la disección de las vesículas seminales.
Contraindicaciones
• Pacientes con Osteoartrosis de cadera, reemplazo de cadera.
• Tamaño de la glándula mayor de 50g.
• Presencia de Lóbulo medio.
• Hemorroides importantes.
Equipo especial (opcional)
• Fotóforo.
• Tractor curvo (Lowsley)
• Tractor recto (Young).
• Separadores de Bookwalter.
Otras concideraciones. . .
• Anestesia general preferiblemente por la posición del paciente.
• Posición de litotomía forzada.
Complicaciones
• Lesión rectal.
• Sangrado.
• Disección prostática incompleta.