Presentación de un Caso y Revisión de la Literatura
Edgar Velasco Zamorano
Miembro Emérito de la Sociedad Colombiana de Urología
Pereira – Colombia
Introducción
Se presenta el caso de una tumoración de la glándula adrenal derecha encontrada sorpresivamente en una laparotomía abdominal abierta, programada para una nefrectomía radical derecha de un hipernefroma descrito en el informe radiológico de un TAC abdominal contrastado, solicitado en el servicio de urgencias para encontrar la causa de un abdomen agudo. Reporte anatomo-patológico final: Feocromocitoma Adrenal (f.a).
El diagnóstico de los desórdenes endocrinológicos de la glándula adrenal (g.a) y de otras glándulas endocrinas del organismo es responsabilidad del clínico. Su ocurrencia es alta pero en realidad no llega a la consulta del urólogo. El estudio y tratamiento médico por lo general corre a cargo del médico internista, el endocrinólogo y el oncólogo. El tratamiento quirúrgico es derivado al cirujano general, al pediátrico, al vascular o al laparoscopista.
En la actualidad la cirugía laparoscópica practicada por cualquier experimentado cirujano se ha convertido en la ideal para el abordaje más fácil y menos traumático de pequeñas o grandes tumoraciones de la g.a y la linfadenectomía correspondiente.
Sospechado cualquier trastorno funcional endocrino por su característica sintomatología se solicitaran los exámenes de laboratorio específicos en sangre venosa y en orina de 24 horas.
Estudios imagenológicos
Los estudios imagenológicos de ultrasonido, urografía excretora, TAC simple y Resonancia Magnética simple, nos revelará solamente el sitio de localización y el tamaño de la lesión. En la actualidad los modernos equipos radiológicos tienden a distinguir de forma no invasiva tumores benignos de los malignos, detectar metástasis regionales o a distancia y descubrir paragangliomas extraadrenales.
En décadas pasadas se debía recurrir a métodos invasivos como eran la arteriografía, la venografía, las biopsias abiertas o percutáneas y las exploraciones quirúrgicas abiertas. El diagnóstico era en un alto porcentaje descubierto por exámenes de autopsias.
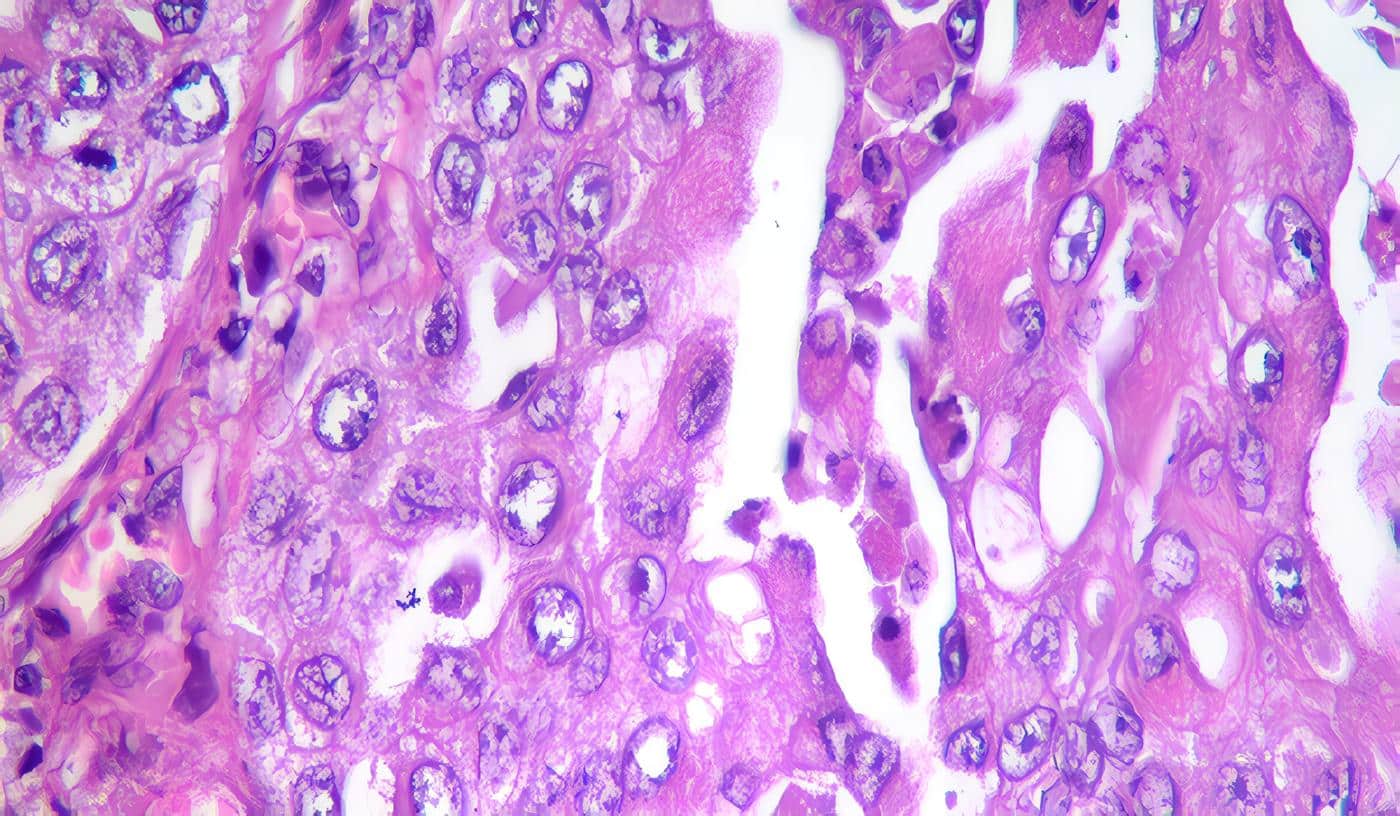
Como en otros tumores endocrinos, el examen histológico solo, utilizando la corriente coloración de hematoxilina-eosina, no puede determinar con seguridad presencia o no de malignidad.
En la solicitud del examen de patología es necesario anexar la sospecha del diagnóstico clínico, ojala con los resultados de laboratorio, rayos X y comportamiento del tumor al tratamiento médico o de observación por un tiempo no menor de seis meses. El patólogo debe practicar estudio inmuno- histoquímicos de tejido tumoral como complemento de un diagnóstico definitivo.
Presentación del Caso
Mujer de 50 años de edad, soltera, sin hijos, ingresa por urgencias con un cuadro abdominal agudo. Diagnóstico probable; oclusión intestinal por vólvulo.
Finalmente un TAC abdominal informa de un tumor maligno (hipernefroma) del polo superior del riñón derecho (figuras 1, 2, 3 y 4).
")
Se solicita interconsulta a urología para practicar nefrectomía radical derecha. Siguiente, se hace resección de un vólvulo intestinal, anastomosis termino-lateral, colecistectomía por inflamación y apendicetomía profiláctica.
Se encuentra tumoración grande que engloba la glándula adrenal derecha y parece introducirse dentro del lóbulo superior renal. Se practica adreno-nefrectomía total y extirpación de ganglios regionales aparentemente no comprometidos.
Al tercer día de estar en la unidad de cuidados intensivos presenta un cuadro séptico por posible peritonitis. Se revisa la cavidad abdominal no encontrando colecciones sanguíneas ni dehicencia de suturas. Se deja la herida abierta y se practican lavados introperitoneales. Gracias al oportuno y dinámico tratamiento de la UCI, la paciente se va progresivamente restableciendo en el lapso de 42 días.
El reporte de patología es enviado 10 días después de haber sido ope rada la paciente. DX: adenocarcinoma adrenal derecho sin compromiso ganglionar. Siete días después llega el informe inmunohistoquímico: Feocromocitoma adrenal positivo para cromogranina, sinoptoficina y vicentina.
Recuperada la paciente se interroga sobre su sintomatología pasada.
Presentaba desde hace 15 años severísima sudoración, cefaleas, taquicardia y crisis de angustia. Fue considerada como síntomas premenopáusicos o psicológicos. Siempre fue vista por médicos generales institucionales. Nunca se le encontró la T.A alta y nunca fue remitida al médico internista. Estos síntomas han desaparecido en la actualidad.
Se revisó el TAC con el radiólogo llegando a la conclusión de que sí podría tratarse de un tumor suprarrenal, maligno por su mayor tamaño, desplazamiento renal, no deformación calicial superior y en una secuencia se observa con mayor nitidez la masa tumoral.
(Lea También: Ultrasonido de la Glándula Adrenal)
Repaso Anatomo-Fisio-Patológico
Como los urólogos también somos clínicos y como la glándula adrenal está íntimamente ligada al riñón, es necesario que no olvidemos su patología y seamos también partícipes en el conocimiento y especialmente en el tratamiento quirúrgico.
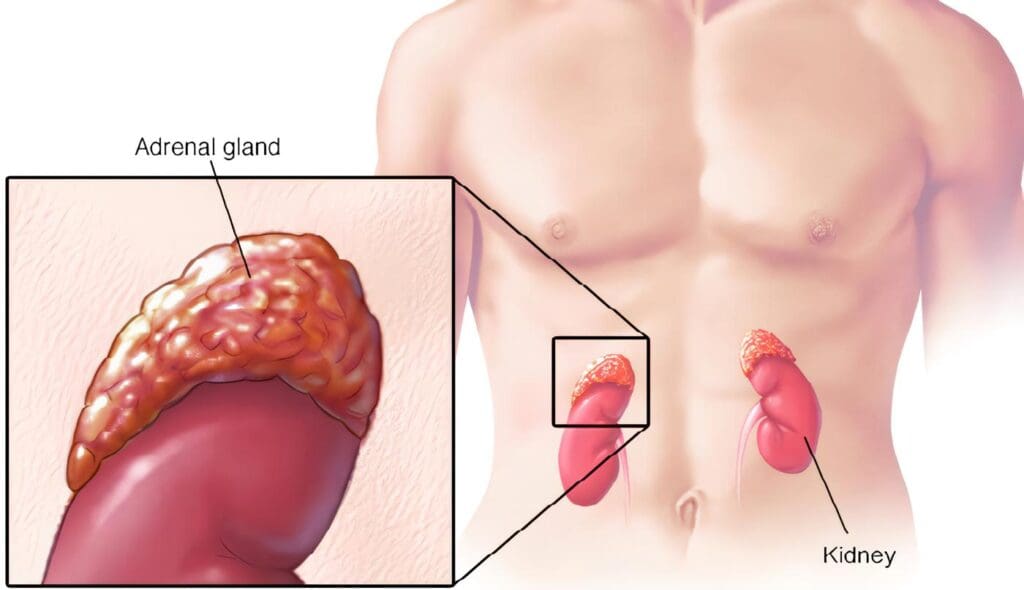
Las glándulas adrenales (g.a) son dos cuerpos pequeños amarillentos, localizados en el espacio perirenal inmediatamente antero-superior al polo superior de los riñones. Son muy vascularizadas y reciben suministro sanguíneo de las arterias suprarrenales superior, media e inferior originadas de las arterias frénica inferior, aorta abdominal y renal, respectivamente.
Las venas medulares emergen del hilio de la glándula para formar las venas suprarrenales, que drenan una a la vena cava inferior en el lado derecho y la otra a la vena renal izquierda.
La g.a. está compuesta de una corteza exterior y una delgada médula interior que representa la décima parte del peso de la glán dula. La corteza a la vez está subdividida en tres zonas; la zona exterior glomerulosa, la zona media fasciculada y la interna reticular.
La corteza produce tres mayores hormonas: el cortisol (glucocorticoide), la aldosterona (mineralocorticoide) y la dehidroepiandrosterona (DHEA), un andrógeno. La medula produce epinefrina (adrenalina), la norepinefrina y la dopamina. Las anormalidades cortico-medulares específicamente producen unos síndromes o enfermedades que vale la pena recordar:
Anormalidades Corticales
El síndrome de Cushing:
Puede presentarse por el exceso de producción de la hormona adreno-corticotropa (ACTH) por parte de la hipófisis (80%), resultando en una hiperplasia cortico-adrenal, por un adenoma adrenal hiperfuncionante (18%), carcinoma adrenal hiperfuncionante (1%) y por producción ectópica de tumores no adrenales (bronquial, timo, tiroides, páncreas): obesidad de la cara y el tronco, mayor grasa intraabdominal, HTA presente o no, acné, oligomenorrea y debilidad generalizada.
Hiperplasia Congénita Adrenal Virilizante
Sindrome de Conn: (aldosteronismo primario):
Por excesiva producción de aldosterona. El 80% por adenoma adrenal unilateral y el 20% por hiperplasia adrenal nodular bilateral. Se caracteriza por H.T.A. benigna, hipokalemia, cefalea, retención de sodio, reducción o ausencia de renina plasmática.
Enfermedad de Addison (Hipoadrenalismo):
Debe destruirse por lo menos el 90% de la glándula para que se produzca. La causa más común en el pasado, era las enfermedades granulomatosas como la TBC y los hongos, en la actualidad la atrofia idiopática de la g.a. es la mayor causa, la hemorragia secundaria a trauma, el stress o la terapia anticoagulante.
Adenomas y Carcinomas Adrenales no Funcionantes Mielolipomas
Anormalidades medulares:
El feocromocitoma.
El neuroblastoma: El neuroblastoma: tiene varios grados de malignidad, desde el más maligno (simpaticogonioma), hasta el benigno ganglio neuroma común en la niñez. Puede ocasionalmente regresar de malignidad a benignidad.
Tumores secundarios: Tumores secundarios: la g.a. es sitio comprometido por metástasis de otros órganos primarios: pulmón, seno, tiroides, colon y melanoma.
Feocromocitoma.
Características generales y sintomáticas
La verdadera incidencia del f.a. es desconocida. Se presenta con igual frecuencia en los hombres y mujeres. Predomina entre los 40 y 50 años de edad. Es a menudo descubierto incidentalmente en exámenes ejecutados con otros propósitos.
El feocromocitoma (f.a) es conocido con los nombres de paraganglioma (P) de la médula adrenal y tumor diez por ciento porque aproximadamente el 10% es bilateral, el 10% es extramedular (paragangliomas), el 10% ocurre en los niños y el 10% son malignos. El f.a. infantil tiene mayor probabilidad de ser bilateral.
El f.a. se deriva en un 90% de las células maduras de la cresta neural en la médula adrenal, llamadas feocromocitos o cromafinomas, también se origina de neuroblastos gangiónicos precursores de la simpaticogonia, lo cual explica la localización extraadrenal de algunos f.a dentro de la cadena ganglionar autónoma simpática a lo largo del eje de la columna vertebral desde el cráneo hasta la región terminal sacra, o también del cuerpo cromafines, tales como el órgano de Zukerkandl (una colección de ganglios simpáticos situados en el origen de la arteria mesentérica inferior), la pelvis o vejiga urinaria.
La simpatogonia se origina del neuroectodermo primitivo o cresta neural, lo cual explica la presencia del F. en varias displacias neuroectodérmicas como la enfermedad de Von Recklinghausen (desorden familiar autosómico dominante, consistente en múltiples neurofibromas, manchas de color café con leche y osteítis quística), la tuberoesclerosis, el síndrome de Sturge-Weber (angiomatosis encéfalo-facial) y la enfermedad de Von Hippel Lindau.
El origen de células parafoliculares tiroideas (precursoras del carcinoma medular del tiroides) del neuroectodermo, puede, de una manera similar explicar la bien conocida relación entre estas lesiones y el f.a, conociéndose dentro del grupo MEN (Neoplasia Múltiple Endocrina).
Ambos los f.a y los paragangliomas secretan norepinefrina, pero solamente los primeros tienen la capacidad de secretar también epinefrina. En los niños ambos secretan solamente noradrenalina.
Los ataques paroxísticos de diaforesis, taquicardia, ansiedad y cefalea se producen por una descarga de catecolaminas al torrente circulatorio. La hipertensión arterial (HA) puede presentarse en ataques o ser persistente, pero también puede no existir.
En algunos estudios no se ha podido demostrar la relación estrecha entre el grado de HA. y los niveles de catecolaminas en la sangre, más aún se ha encontrado hipotensión arterial en algunos pacientes, debido muy posiblemente a la desensibilización que sufre el sistema cardiovascular a las catecolaminas, por lo tanto a mayor edad menor es la sintomatología y sí se añade las enfermedades sobreagregadas del viejo, el médico tendría más posibilidades de equivocarse en el diagnóstico clínico.
En pacientes a quienes no se les ha diagnosticado el f.a. con el correr de los años pueden llegar a presentar edema pulmonar agudo por falla ventricular izquierda de miocardiopatía dilatada o por causa no cardiogénica. Puede presentar hemorragias, infartos y embolias cerebrales, también anorma lidades metabólicas como diabetes e hipercalcemia.
Raramente, la ruptura espontánea de un f.a. o un p. puede ser la causa de letal hemorragia retroperitoneal manifestándose como un abdomen agudo. Los p. tienen un curso más agresivo que su contraparte, aproximadamente el 20% al 42% de p. metastatisan, comparados con solamente el 2% de los f.a. La metástasis se hace por vía linfática y hematógena, siendo los ganglios linfáticos regionales, el hueso, el hígado y los pulmones.
A Causa de que los p. benignos y malignos tienen una idéntica apariencia histológica, su comportamiento clínico (por ejemplo enfermedad recurrente o metástasis) es el mejor predictor del pronóstico.