Caso Clínico Interinstitucional
Julián D. Martínez M., M.D., Luis G. Guevara, M.D., Diego Gómez, M.D., Martín A. Garzón, M.D., Mario H. Rey T., M.D., Juan C. Molano, M.D., Juan C. Marulanda, M.D., Unidad De Gastroenterología,
Hospital Universitario De La Samaritana. Bogotá, D.C.
Se trata de un varón de 23 años, soltero, de ocupación electricista, con escolaridad completa, quien consulta a esta institución por primera vez en junio de 2000, por astenia, prurito generalizado, anorexia e ictericia mucocutánea de cuatro semanas de evolución, sin informar otros síntomas.
Como antecedente informó, desde la edad de ocho años, de múltiples episodios de ictericia, coluria y prurito, de tres a cinco meses de duración y con autorresolución de los mismos; se le realizaron múltiples exámenes paraclínicos, entre ellos una biopsia hepática durante un periodo anictérico, a los 16 años de edad, que fue informada como normal, sin obtener un diagnóstico definitivo.
No se conocieron otros antecedentes personales o familiares de importancia. Al examen físico se observó un paciente en buen estado general, con ictericia mucocutánea y signos de rascado en dorso y extremidades, como únicos hallazgos positivos.
Los exámenes de laboratorio de hemograma:
Extendido de sangre periférica, recuento de reticulocitos y pruebas de coagulación fueron normales; la bilirrubina total (BT) fue de 12 mg/dl, la bilirrubina directa (BD) de 8,0 mg/dl, la fosfatasa alcalina (FA) de 960 U, y las aminotransferasas normales; la albúmina sérica, la creatinina, el nitrógeno ureico, y el ultrasonido abdominal fueron normales; las serologías para hepatitis A, B, C fueron negativas, como también la prueba de Coombs directa.
La revisión de exámenes de laboratorios realizados durante episodios anteriores, mostró un patrón de ictericia colestásica, dos ecografías hepatobiliares normales y serologías para hepatitis virales no reactivas.
Se inició manejo con suplencia de vitaminas liposolubles y ácido ursodesoxicólico, 600 mg/día.
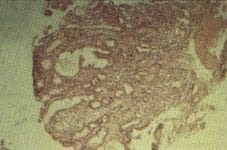
Los ANAS, AMAS fueron negativos; la ceruloplasmina y la ferritina séricas fueron normales. Se realizó una colangiopancreatografía retrógrada endoscópica (CPRE) la cual mostró la vesícula biliar y las vías biliares intra y extrahepática normales, y un conducto de Wirsung normal (Figuras 1 y 2).
[enc_su_row][enc_su_column size=”1/2″]

Figura 1. CPRE normal.

[/enc_su_column] [enc_su_column size=”1/2″]

Figura 2. CPRE normal.
[/enc_su_column] [/enc_su_row]
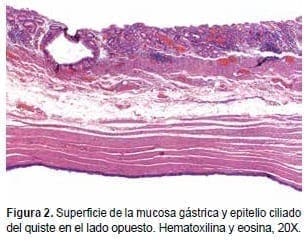
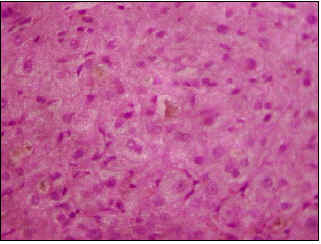
En agosto de 2000, se realizó una laparoscopia que mostró un hígado de aspecto colestásico, de tamaño, forma, contornos y superficie usuales. La biopsia hepática por trucut fue informada como: colestasis sin actividad necro-inflamatoria (Figuras 3-5).
Figura 3. Colestasis intrahepática (X10).

[enc_su_row][enc_su_column size=”1/2″]

Figura 4. Colestasis intrahepática (X40).
[/enc_su_column] [enc_su_column size=”1/2″]
Figura 5. Colestasis intrahepática (X100).

[/enc_su_column] [/enc_su_row]
En un control posterior, a las cinco semanas, se observó desaparición de la ictericia.
Comentarios
La colestasis intrahepática benigna recurrente (BRIC, de su sigla en inglés) es un síndrome caracterizado por múltiples episodios de prurito e ictericia.
Es más frecuentes en los hombres y se inicia entre los 10 y 30 años de edad. Los ataques tienen una duración individual que varía entre semanas y meses (con duración promedio de 3 meses) y se resuelven de manera espontánea y no conllevan a disfunción hepatocelular progresiva, fibrosis o cirrosis (1,2).
Los criterios diagnósticos incluyen:
- El antecedente de, al menos, dos episodios de ictericia con un intervalo libre de síntomas de meses o años de duración (en promedio de dos años );
- Pruebas de laboratorio consistentes con colestasis: elevación de fosfatasa alcalina, al menos el doble del límite superior normal (aunque en algunos pacientes se han registrado elevaciones de más de 40 veces), hiperbilirrubinemia conjugada con 10 mg/dl como cifra promedio, gamma GT con mínimas elevaciones, aminotransferasas en niveles normales o mínimamente elevadas y ácidos biliares elevados;
- Presencia de prurito secundario a colestasis (3-5), aunque hasta el 25% de los pacientes no lo informan. Otros síntomas asociados son astenia, adinamia, náuseas, vómitos, anorexia, cefalea, fiebre y dolor en el hipocondrio derecho en la mitad de los afectados;
- Ausencia de otros factores conocidos como causantes de colestasis, como medicamentos, anovulatorios y embarazo;
- Las vías biliares intra y extrahepática son de morfología normal demostrada mediante colangiopancreatografía retrógrada endoscópica (CPRE) (2); y,
- Una biopsia hepática que muestre colestasis centrilobular. La bilis se puede observar también en los canalículos, los hepatocitos y las células de Kupffer; otros hallazgos, menos frecuentes, incluyen degeneración hepatocelular pericentral, necrosis hepatocitaria y escasas áreas de inflamación (5); estos cambios se resuelven totalmente en los períodos intercríticos.
Se han informado más de un centenar de casos en todo el mundo y cerca de la mitad de estos tienen antecedentes familiares de colestasis.
La predisposición familiar es notoria en el BRIC; los gemelos son afectados más frecuentemente y se han informados múltiples casos de madres e hijos afectados (6).
Los estudios genéticos han demostrado un gen mutante en el brazo largo del cromosoma 18 (18q21-22) y es denominado como FIC-1 (familial intrahepatic cholestasis); dicho gen se localiza en múltiples epitelios (gastrointestinal, pancreático, hepático).
En los pacientes con BRIC, se informan dos mutaciones (7); el producto del FIC-1 en una proteína ATPasa tipo P, la cual participa en el transporte de cationes como cobre, calcio, sodio y potasio.
Además, una subfamilia de esta ATPasa ayuda a dar una correcta posición en las membranas celulares a los aminofosfolípidos como la fostatidilserina (PS), la fosfatidiletanolamina (PE), la fosfatidilcolina (PC) y la esfingomielina (SM).
Por tanto, un defecto en el funcionamiento de esta ATPasa acarreará una posición errónea de los lípidos de la membrana celular, modificando su fluidez y alterando procesos de transporte celular mediado por proteínas y los procesos de exocitosis, lo cual daría lugar a una secreción reducida de ácidos biliares en el hígado.
La mutación en el páncreas produciría una secreción exocrina defectuosa y, en el íleon, una notoria disminución en la reabsorción de sales biliares, lo que explicaría la pérdida aumentada de estas sales observada en los pacientes con BRIC (8,9).
El diagnóstico diferencial incluye múltiples causas de ictericia y colestasis intrahepática. Una adecuada historia clínica y los estudios de laboratorios, excluyen toxicidad por medicamentos, hepatitis infecciosas, enfermedad hepática alcohólica y colestasis del embarazo.
La CPRE descarta la presencia de causas obstructivas y la histología hepática excluye la cirrosis biliar primaria, los granulomas, etc. (2).
No hay tratamiento específico para los episodios de BRIC:
Tampoco para prevenirlos ni para acortar su duración. Se han empleado múltiples medicamentos en el manejo del prurito; los antihistamínicos han mostrado resultados modestos, las resinas quelantes de la sales biliares en el lumen intestinal, como la colestiramina, tampoco demuestran grandes resultados terapéutico, y así mismo sucede con los corticoesteroides.
Sustancias como el fenobarbital, que induce la síntesis de citocromos y reduce la excreción urinaria y la toxicidad de los ácidos biliares, también se han utilizado con resultados dispares; la rifampicina, que además de reducir la flora intestinal (inhibiendo de paso la conversión de sales biliares primarias a secundarias de mayor toxicidad) es un inductor de varios citocromos, ha demostrado mejoría del prurito y disminución de los niveles de bilirrubina y fosfatasa alcalina.
El SAME (S-adenosilmetionina), al repletar los hepatocitos de glutatión, aumenta el metabolismo de los fosfolípidos y la fluidez de las membranas celulares; no ha demostrado mejoría del prurito o de las anormalidadesbioquímicas del BRIC.
El ácido ursodesoxicólico (UDCA), cuyos efectos principales consisten en desplazar las sales biliares endógenas tóxicas de la circulación enterohepática (inhibición competitiva de la absorción de sales biliares endógenas por el íleon), compite por receptores en los organelos celulares en donde los ácidos biliares tóxicos pueden ocasionar daños y, como consecuencia, mejora la función excretora del hígado, ejerce un papel protector sobre las membranas celulares (mitocondriales, canaliculares).
Previene la citólisis y la apoptosis inducidas por los ácidos biliares, ejerce un efecto antioxidante que previene la peroxidación y es un inmunomodulador que disminuye la liberación de citocinas (interleucinas 2 y 4, y gammainterferón), ha desmotrado, en múltiples estudios, su capacidad para disminuir la colestasis en los pacientes con BRIC y es, hasta ahora, la sustancia recomendada en su tratamiento (2,10-14).
Referencias
- 1. Summerskill WHJ. The syndrome of bening recurrent cholestasis. Am J Med 1965;38:298-305.
- 2. Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clinics in Liver Disease 1999;3(3):1-19.
- 3. Tygstrup N, Jensen B. Intermittent intrahepatic cholestasis of unknown etiology in five young males. Acta Medica Scandinavica 1969;185:523-30.
- 4. De Pagter AGF, Van Berge, Henegouwen GP, Bokkel-Huinnuk J, et al. Familial bening recurrent cholestasis. Gastroenterology 1976;71:202-07.
- 5. Renard R, Geubel AP, Benhamou JP. Bening recurrent intrahepatic cholestasis. J Clin Gastroenterol 1989;11:546-51.
- 6. Da Silva LC, Brito T. Bening recurrent intrahepatic cholestasis in two brothers. Ann Intern Med 1965;65 :330-41.
- 7. Oude E, Van Berge. Cracking the genetic code for bening recurrent and progressive familial intrahepatic cholestasis. J Hepatl 1998;29:317-20.
- 8. Minuk GY, Shaffer EA. Bening recurrent intrahepatic cholestasis, Evidence for an intrinsic abnormality in hepatocite secretion. Gastroenterology 1987;93:1187-93.
- 9. Bijleveld CM, Vonk RJ, Kuipers F, Havinga R, et al. Bening recurrent intrahepatic cholestasis: altered bile acid metabolism. Gastroenterology 1989;97:427-32.
- 10. Bergasa NV, Jones EA. The pruritus of cholestasis. Sem Liver Dis 1993;13:319-27.
- 11. Stiehl A, Benz C, Sauer P. Mecanismo de la acción hepatoprotectora de las sales biliares en las hepatopatías. Clínicas de Gastroenterología de Norteamérica. 1999;1:213-28.
- 12. Maggiore G. Efficacy of ursodeoxycholic acid in preventing cholestasic episodes in a patient with bening recurrent intrahepatic cholestasis. Hepatology 1992;16:504.
- 13. Bircher J. Treatment of patients with bening recurrent intrahepatic cholestasis. Hepatology 1989;10:1030-32.
- 14. Guevara LG. Colestasis. Terapéutica en hepatología, 1a. Edición. Asociación Colombiana de Hepatología, Bogotá D.C, 2001:196-205.