La patogénesis de la ateroesclerosis en no diabéticos tiene recientes y muy completas revisiones y siempre se inician por el análisis de la disfunción endotelial23.
Esta condición también está presente en las arterias de individuos con resistencia a la insulina24, 25
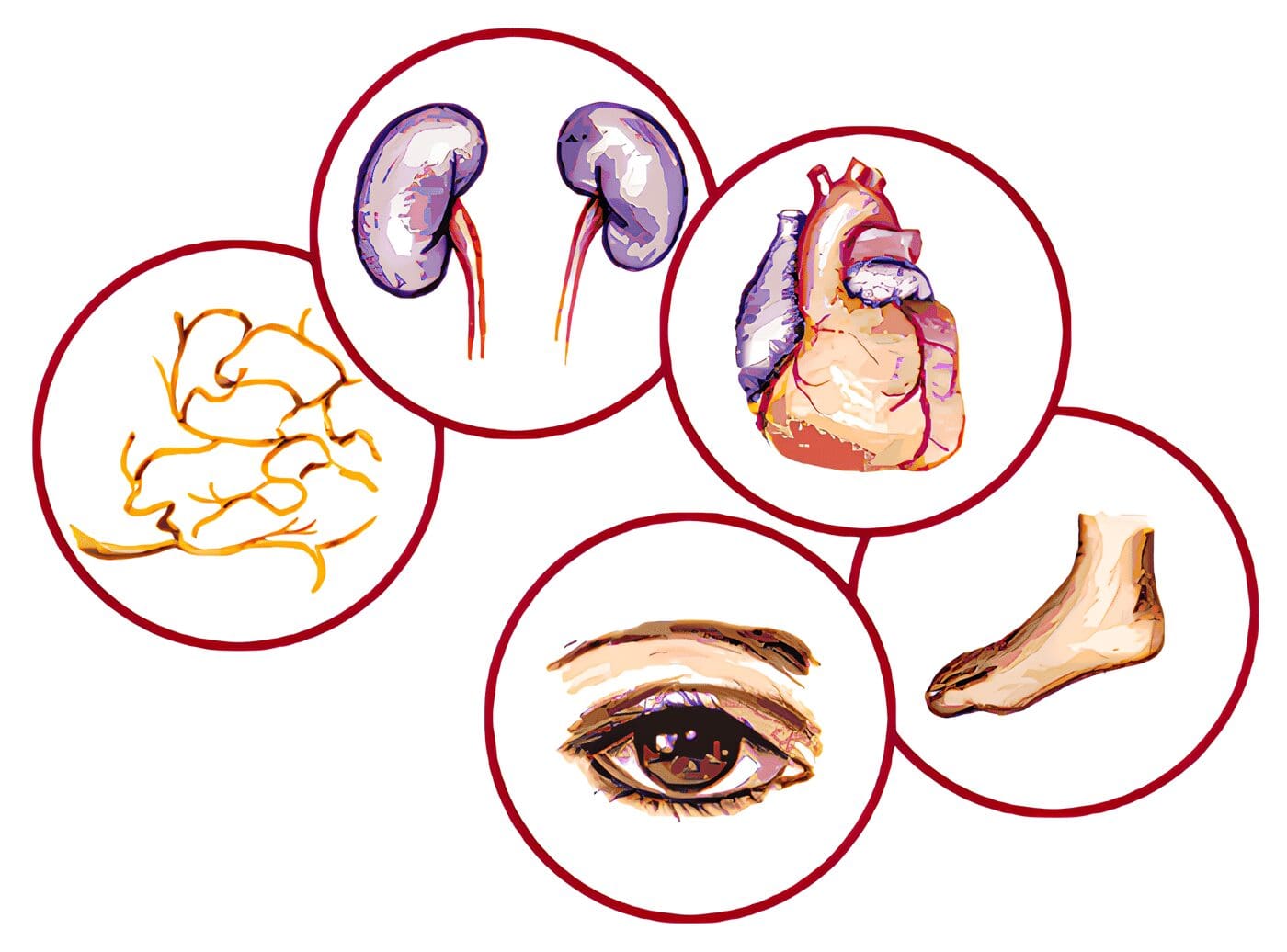
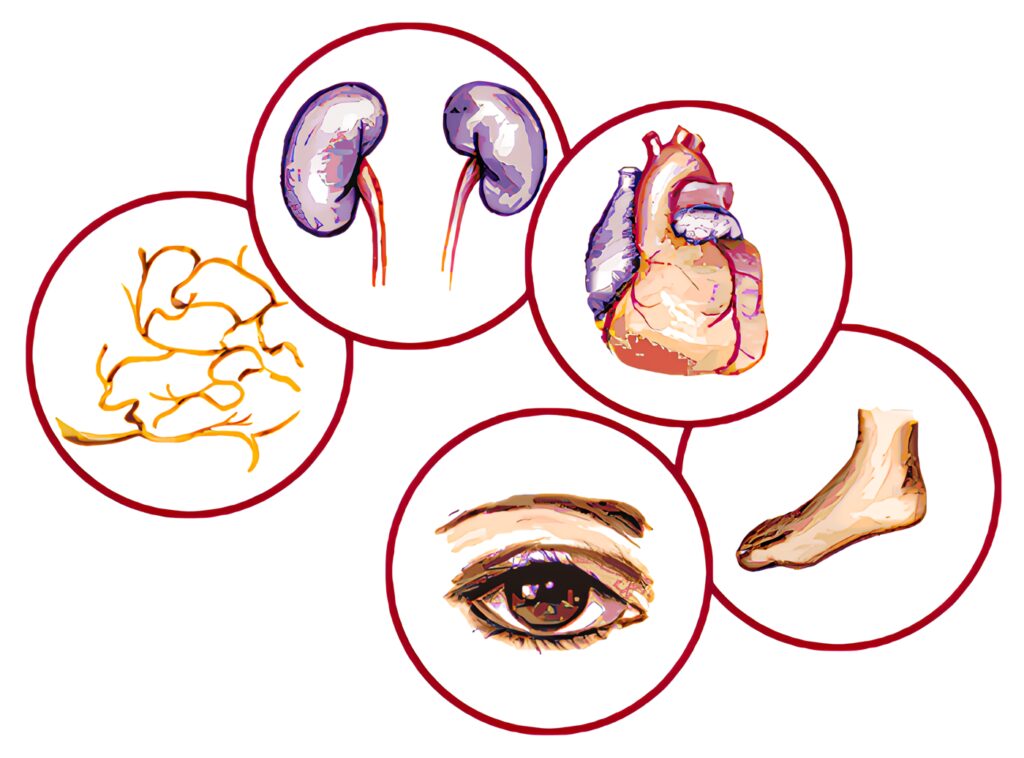
La enfermedad micro vascular diabetes/específica en la retina, en el glomérulo renal y en los vasa nervorum, tienen mecanismos fisiopatológicos similares.
En el comienzo de la enfermedad, la hiperglicemia intracelular produce en el humano anormalidades en el flujo sanguíneo26.
Estas alteraciones conllevan una disminución en la actividad de los factores vasodilatadores intracelulares, tales como el óxido nítrico (NO) y un aumento en la vasoconstricción por incremento de la angiotensina II y la endotelina-1, todo lo cual viene acompañado con el incremento en la elaboración de los factores de permeabilidad, tales como el factor de crecimiento vascular endotelial (VEGF).
Al propio tiempo aparecen las modificaciones cuantitativas y cualitativas de la matriz extracelular, que dejan una hiperpermeabilidad vascular irreversible.
Viene entonces la pérdida de células de la estructura micro vascular, dentro del programa de apoptosis, todo lo cual conduce a la oclusión capilar y a la sobreproducción de la matriz extracelular.
En este momento, es cuando los factores transformadores de crecimiento (TGF-b) participan en el depósito de proteínas plasmáticas PAS +.
Finalmente, la hiperglicemia puede conducir a una disminución de los factores tróficos endoteliales y de la neurona.
Todos estos cambios llevan al edema, a la isquemia y a la hipoxia inducida por la neovascularización en la retina que corren parejas con la expansión de la matriz mesangial, la glomérulo esclerosis y la degeneración axonal de los nervios periféricos.
Pero ¿cuáles son los mecanismos por los cuales la hiperglicemia induce el daño microvascular?
Cuatro hipótesis fundamentales acerca de cómo la hiperglicemia causa las complicaciones vasculares han generado un número importante de estudios epidemiológicos, químicos y biomoleculares.
Los tres modelos de complicación micro vascular precoz, antes de haberse establecido la diabetes, y analizadas a través de estudios epidemiológicos son:
- la neuropatía
- la retinopatía
- la micro-proteinuria renal (preludio de la nefropatía diabética).
Los mecanismos por los cuales la hiperglicemia induce el daño vascular están basados en cuatro hipótesis27:
- Aumento en la vía del flujo de los polioles
- Aumento de los productos de glicosilación avanzadas (AGE)
- Activación de las isoformas de la proteína-cinasa C (PKC)
- Aumento en el flujo de la vía de la hexosamina

La aldosa reductasa (Aditol: NAD(p)+1/oxidorreductasa) es la primera enzima en la vía de los polioles.
Es citosólica y cataliza el NADPH dependiente de la reducción de una amplia variedad de compuestos carbonilos que incluyen la glucosa.
La aldosa reductasa tiene muy baja afinidad con la glucosa y hay concentraciones normales de glucosa en personas no diabéticas.
El metabolismo de la glucosa por esta vía es de muy bajo porcentaje. Al aumentar la glucosa intracelular se aumenta la conversión del poli alcohol sorbitol con la correspondiente disminución de NADPH.
Durante la hiperglicemia el flujo de esta vía es aproximadamente de 33% del total de la glucosa en uso y del 11% en los glóbulos rojos humanos.
El efecto de la hiperglicemia es el aumento del flujo de la vía de los polioles que incluye el sorbitol y, que no se difunde fácilmente a través de las membranas por lo cual se ha sugerido que da lugar a un daño osmótico micro vascular.
Sin embargo está lejos de dar lugar a una alteración osmótica definitiva.
La hipótesis más reciente es que la oxidación del sorbitol por el NAD aumenta la relación NADH:
NAD+ inhibiendo la actividad de la dehidrogenasa liceraldehído/trifosfato (GADPH) y aumentando la concentración de triosafosfato28. Al elevarse las concentraciones de triosafosfato se aumenta la formación de metilglioxal, un precursor de los AGEs y del diacilglicerol (DAG) que activa el PKC.
Los estudios de la inhibición de la vía de los polioles ha dado resultados inconsistentes. El único efecto positivo en un estudio multicéntrico controlado demostró que el zenarest es un potente inhibidor de la aldosa reductasa29.
Lea También: La Mitocondria y la Producción de Superóxido, Mecanismos Biomoleculares y Patogénicos
La segunda hipótesis plantea el aumento en la formación de productos finales de glicosilación avanzada (AGEs):
Estos productos AGEs se encuentran en mayores cantidades en los vasos retinianos y glomerulares de los diabéticos30.
Se forman a partir de grupos carbonilos derivados de la glucosa que se generan intracelularmente, siendo la hiperglicemia el primer evento para su formación intra o extracelular31.
Los AGEs provienen de una auto-oxidación intracelular de glucosa a glioxal por descomposición del producto Amadori (glucosa derivada 1amino/1 de oxifructuosa) o a 3/de oxiglucosona, posiblemente acelerada por una amadoriasa y por fragmentación del gliceraldehído 3/fosfato y fosfato de dehidro-oxiacetona a metilglioxal y deoxiglucosona, reaccionando con amino grupos intracelulares y extracelulares para formar los AGEs.
El metiglioxal y el glioxal son detoxificados por el sistema glioxalasa.
La producción intracelular de precursores AGE daña las células blanco por tres mecanismos:
- Proteínas intracelulares modificadas por los AGEs alteran su función
- Los componentes de la matriz extracelular modificada por los precursores de los AGEs interactúa con otros componentes de la matriz y sus receptores proteicos integrinas
- Las proteínas plasmáticas modificadas por los precursores de los AGEs se unen a sus receptores en las células endoteliales, en las células mesangiales y en los macrófagos induciendo la producción de especies reactivas de oxígeno (ROS).
La formación de AGEs altera las propiedades funcionales de varias moléculas importantes de la matriz endotelial. Un tipo de colágeno intermolecular al ligarse con los AGEs induce la expansión del paquete molecular32.
La formación de AGEs en la matriz extracelular no solamente interfiere en las interacciones de la misma matriz sino que interfiere entre las interacciones célula-matriz, como sucede en el caso de la modificación del colágeno tipo IV cuyos dominios de ligadura disminuyen la adhesión de las células endoteliales33.
Diversas proteínas que se unen con los AGEs se han identificado que incluyen la galectina/3, el receptor barrendero tipo II del macrófago y el receptor RAGE34
Consistente con este concepto de bloqueo de un receptor RAGE el cual es un miembro de la superfamilia de las inmunoglobulinas que inhibe el desarrollo de la nefropatía, aumenta, promueve la reparación de heridas y media en la traducción y generación de especies de oxígeno reactivo, activando el factor NF/KB35.
La tercera hipótesis Del efecto de la hiperglicemia
Sobre el daño vascular es la activación de la proteínquinasa C. (PKC) La familia de la PKC comprende quince isoformas, nueve de las cuales son activadas por el segundo mensajero de los lípidos, el diacil glicerol (DAG).
La hiperglicemia intracelular aumenta el DAG en las células microvasculares de la retina y en el glomérulo renal de los animales diabéticos. La síntesis del DAG se aumenta a través de la reducción del fosfato de hidrooxiacetona a glicerol 3/fosfato.
Las isoformas b y d se activan primariamente a través de la hiperglicemia y también indirectamente a través de los receptores AGEs y por incremento de la vía de los polioles, aumentando a su vez las especies reactivas de oxígeno (ROS). La activación de las isoformas PCK/b media en las anormalidades del flujo retinal y renal, posiblemente por disminución en la producción de óxido nítrico y por aumento de la actividad de la endotelina/136.
También la activación del PKC está implicada en la disminución glomerular de óxido nítrico en el músculo liso, lo cual está inducido por la hiperglicemia. La activación del PKC también inhibe la expresión del mRNA estimulada por la insulina, para la síntesis endotelial del óxido nítrico (eNOS)37.
La hiperglicemia a su vez aumenta la endotelina 1 estimulada por la MAP/quinasa en el glomérulo y en el mesangio activada por las isoformas PCK. La activación del PCK por la hiperglicemia también induce la expresión de la permeabilidad aumentando el VEG del músculo liso38.
La hiperglicemia induce anormalidades en el flujo sanguíneo en la permeabilidad endotelial.
El PKC contribuye al aumento de la matriz microvascular por inducción en la expresión del TGF/b1 la fibrinonectina y el colágeno39.
El efecto es mediado posiblemente por la inhibición en la producción del ácido nítrico por el PKC. El PCK activado por la hiperglicemia induce la mayor expresión del factor activador inhibidor/1 del activador del plasminógeno PAI/140
El tratamiento experimental con inhibidores específicos PKC/b reduce la actividad del PKC en la retina y en el glomérulo renal de los animales en experimentación al mejorar el tiempo de circulación, la filtración glomerular y la corrección de la excreción de la albúmina urinaria, con las mismas isoformas inhibidoras en ratones DB/DB al inhibir el PKC se mejoró la expansión acelerada del mesangio.
La última hipótesis es el aumento del flujo a través de la vía de la hexosamina. El exceso de glucosa dentro de la vía de la hexosamina causa diversas complicaciones vasculares en los animales de experimentación.
En la vía normal de la glicólisis se producen substratos que requieren UDPN/UDP N acetil glucosamina que se requiere para la síntesis del proteoglicano, con la formación de glicoproteínas unidas al oxígeno.
Al inhibir esta enzima que convierte la glucosa en la glucosamina mediante una glutamino fructuosa amidotransferasa (GFAT), puede bloquearse la hiperglicemia que ha intervenido en los factores de trascripción ya mencionados TGF/a, TGF/b1 y PAI/141.
Esta vía es muy importante en el papel de la hiperglicemia que induce la resistencia de la insulina, a su vez inducida por las grasas.
La vía de la hexosamina y la hiperglicemia consecutiva induce también cambios en la trascripción del gen para el PAI/1.
En síntesis, la activación de la vía de la hexosamina por la hiperglicemia puede resultar en diversos cambios tanto en la expresión del gen como en la función de las moléculas proteicas que forman parte de toda la cadena de eventos que llevan a las complicaciones vasculares de la diabetes.