Diana García de Olarte
Pediatra, Doctor en Ciencias Básicas Biomédicaz
Profesor Facultad de Medicina, Universidad de Antioquia
Hace cinco décadas, el timo era considerado un órgano misterioso que se irradiaba como tratamiento para el estridor respiratorio, el trasplante de órganos era ciencia ficción, la determinación de las gammaglobulinas era un esfuerzo sofisticado, no se conocía la función de los linfocitos y las inmunodeficiencias no se habían descubierto.
Desde entonces, se ha generado abundante información inmunológica con el trabajo combinado de muchos científicos.
Puede considerarse que la Inmunología moderna se inició en 1952, cuando el coronel Ogden Bruton (1), estableció la descripción y el estudio de las inmunodeficiencias primarias (IDP) con su notable reporte, en el cual reseñó el cuadro clínico de un niño de ocho años, quien desde los cuatro y medio años de edad había sufrido 19 episodios de sepsis, la mayoría producidas por Streptococccus pneumoniae.
Las inmunizaciones con vacunas de Streptococcus pneumoniae autógeno o mezclas de polisacáridos tipo específicos no indujeron anticuerpos (acs). Una prueba de Shick positiva, indicó la ausencia de acs contra la toxina diftérica, también el niño falló en responder a la vacuna tifoidica.
El paciente tuvo parotiditis epidémica en tres ocasiones y no desarrolló acs fijadores del complemento contra el virus de las paperas.
El análisis del suero utilizando electroforesis de proteínas, técnica que sólo había sido aplicada recientemente en los análisis clínicos, reveló la ausencia del pico de las g globulinas, aunque las fracciones de proteínas totales y albúmina fueron normales.
Bruton completó su extraordinaria investigación clínica instituyendo un tratamiento con inyecciones intramusculares de globulina sérica inmune humana (GSIH). Él observó la presentación de un pico de g globulinas en el suero del niño y su desaparición después de seis semanas; con inyecciones mensuales el paciente no tuvo sepsis durante 14 meses.
Estas dosis y el intervalo para la administración de GSIH:
Se utilizaron para tratar las enfermedades por deficiencia de acs en los siguientes treinta años. Estos hallazgos llevaron a Bruton a acuñar el término agammaglobulinemia y a deducir que había una conexión entre la ausencia de inmunoglobulinas (Igs) y la susceptibilidad a las infecciones.
Pero seguramente, lo más importante del informe original de Bruton, fue que inspiró a un gran grupo de investigadores para explorar pacientes con deficiencias de acs.
Así, investigaciones posteriores conducidas por Charles Janeway 1953 (2) y Robert Good 1956, (3) demostraron que muchos niños estaban afectados en forma similar al descrito por Bruton y probaron que la Agammaglobulinemia, era un trastorno que se heredaba con un patrón recesivo ligado al cromosoma X.
Pocos años después se identificaron muchos pacientes en todo el mundo, no sólo con Agammaglobulinemia ligada al cromosoma X (ALX), sino con otras anormalidades en la síntesis de Igs (Good y cols, 1962) (4).
Sin embargo, el reconocimiento de diferentes formas de deficiencias de acs fue un proceso gradual y en los primeros años de la década de los setentas, se efectuó una clasificación que fue aceptada como se conoce hoy (Cooper y cols. 1973) (5).
En los años cincuentas, la relación entre los linfocitos y las células plasmáticas era aún controvertida, y los estudios realizados en ALX, fueron esenciales en este análisis (Good y cols. 1956) (3).
Un progreso rápido en las descripciones fenotípicas de varios síndromes, ocurrió con la transformación de la ciencia de la inmunología, desde su enfoque en la serología, a las disciplinas de inmunoquímica e inmunología celular. Robert Good (6) y Henry Kunkel (7), fueron líderes en estas áreas y lo lograron en parte, porque fueron capaces de reconocer el valor de las enfermedades por inmunodeficiencia y por autoinmunidad, como experimentos de la naturaleza (8-10).
Otro acercamiento basado en la manipulación de embriones de ratón in vitro:
Para suprimir o inactivar por recombinación homóloga un gen específico, generó los ratones Knock-out. En ellos se pueden observar los efectos producidos por la ausencia de genes particulares. Este instrumento se convirtió en uno de los más poderosos empleados en la biología moderna.
Las observaciones sobre las respuestas de hipersensibilidad retardada (HR) intactas que presentan los pacientes con Agammaglobulinemia (Good y cols. 1955) (11) pusieron en escena el reconocimiento de los dos compartimentos del sistema inmune (SI).
Con el descubrimiento de la aparición de una deficiencia en la producción de acs en pollos, luego de la remoción temprana de la bursa de fabricius (Cooper y cols. 1996 (12), Cooper y cols. 1969) (13), llevaron a cabo los experimentos básicos en los cuales demostraron el desarrollo independiente de los sistemas del timo y de la bursa de fabricius, responsables de la inmunidad celular y humoral, respectivamente.
La asociación de aplasia tímica con hipoparatiroidismo congénito la descubrió inicialmente Lodbell en 1959 (14), pero sólo fue reconocido como síndrome en 1965 cuando Angelo DiGeorge (15), describió un grupo de niños con ausencia congénita del timo y de la paratiroides; en 1979 Conley y cols. (16), informaron sobre la dismorfia facial y los defectos cardíacos que le acompañaban.
Así, la anomalía de DiGeorge y la ALX, fueron los experimentos de la naturaleza ahora replicados por la timectomía o la bursectomía en los pollos.
El modelo emergente de dos compartimientos para el desarrollo linfoide, se adaptó fácilmente a los casos nuevos descritos de inmunodeficiencia combinada (IC), en los cuales se postuló un defecto en las células madres linfoides.
La restauración de la inmunidad funcional:
En un paciente con inmunodeficiencia severa combinada (IDSC) con trasplante de médula ósea (mo) histocompatible por Gatti y cols. 1968) (17) y en un paciente con síndrome de DiGeorge, con trasplante de timo fetal por Cleveland y cols. 1968 (18), confirmó en forma elegante este modelo.
En los primeros años de la década de los setentas fue posible realizar la identificación de las células B y T, como poblaciones separadas de la sangre y de los linfocitos tisulares, y los estudios de pacientes con Agammaglobulimenia, Cooper y cols. 1971 (19), Seigal y cols. 1971 (20), ayudaron a delinear la diferenciación que existe en el tejido linfoide.
El progreso de los sistemas de cultivo permitió la diferenciación de los linfocitos B a células plasmáticas secretantes de Igs e hizo posible el delineamiento de los defectos funcionales de las células T y B (WU y cols. 1973) (21), de otro lado, el comprobar que los pacientes con ALX tenían un número normal de células pre-B en la mo, contribuyó al proceso de identificación del locus del defecto de diferenciación y en la confirmación del papel que tienen estas células en el desarrollo del linaje B (Pearl y cols. 1978) (22).
Naor y cols. en 1969 (23), describieron por primera vez la ausencia de células que fijan antígeno (ag) en pacientes con ALX; e investigaciones posteriores (Sideras y Smith, 1995 (24), empleando anti-Igs marcadas con fluorocromos, demostraron en forma concluyente el defecto en la diferenciación de los linfocitos B.
En los años ochentas, con las posibilidades de identificar y de manipular in vitro los linfocitos T y B, se adquirió una mayor información sobre las patogénesis de las IDP.
En la última década, los avances de la biología molecular:
Han brindado una mayor comprensión de los procesos que regulan el crecimiento, la diferenciación, la comunicación y las funciones efectoras de los componentes del SI. Estudios recientes en estas enfermedades (quizás más que en cualquier otro tipo de patologías) han revelado el poder de la genética molecular moderna, en términos moleculares precisos.
Royer Pokora y cols. en 1986 (25), clonaron el gen de la gp91 phox de la NADPH oxidasa de los neutrófilos (poly) y lo denominaron gen CYBB; está localizado en la región Xp21.1 del brazo corto del cromosoma X, lo que constituyó la primera caracterización molecular de una IDP. Hasta 1993, sólo se conocía un grupo pequeño de genes asociados con IDP. Vetrie y cols. 1993 (26) y Tsukada y cols. 1993 (27), utilizando dos acercamientos diferentes, descubrieron el gen afectado en la ALX que codifica una nueva tirosina kinasa citoplasmática de los linfocitos B, gen BTK (Bruton tirosina kinasa). Graf y cols. en 1992 (28), clonaron el gen que codifica el CD40 ligando en la inmunodeficiencia ligada al X con incremento de la IgM.
Después de estos informes, el universo de la genética molecular se ha expandido en forma monumental, al punto que cada mes, se reporta la identificación de un nuevo gen asociado a una deficiencia inmune. Esta nueva concepción de las bases inmunológicas y de la patogénesis de las IDP, permite que ellas se reconozcan como enfermedades genéticas.
El número total de genes en los humanos está cercano a los 100.000:
Los comprometidos en el desarrollo y función de los leucocitos son alrededor de 10.000, y es posible que 1.000 genes únicos estén relacionados a las IDP, lo cual indica que estas entidades pueden exceder en diez veces más al número identificado actualmente (cien). La verdadera frecuencia de estos genes en diferentes poblaciones, es aún aproximada.
Los síndromes por inmunodeficiencia severa combinada, representan muchos modelos para el estudio de las células T y en algunos, para la diferenciación de los linfocitos B (LB) y de las células asesinas naturales (NK). Un progreso significativo se ha alcanzado en los últimos años con la identificación de los genes relacionados con las IDSC, lo cual ha aportado una visión sobre el papel de las moléculas claves, comprometidas en la proliferación o diferenciación de los precursores de los linfocitos.
La IDSC representa la forma más severa de las IDP. Estas enfermedades comparten la característica de un profundo bloqueo en la diferenciación de las células T y B y son letales en el primer año de vida. Debido a la ausencia de la inmunidad mediada por los linfocitos T, las infecciones por oportunistas algunas veces causadas por vacunas vivas, asociadas a desnutrición profunda, son las responsables de la muerte. Hasta el presente se han reconocido diferentes formas y se han agrupado de acuerdo al patrón de herencia, al fenotipo y a la identificación de los genes mutados.
Schwarz y cols. en 1991 (29), encontraron que hay una recombinación V(D)J anormal en pacientes con IDSC con linfocitos T- B- .
Cavazzana y cols. en 1993 (30), descubrieron que las células T- B- de muchos pacientes tenían una sensibilidad excesiva a la radiación ionizante. En los pacientes que no presentan esta alteración, Schwarz y cols. 1996 (31), hallaron mutaciones en los genes Rag1, Rag2 o en ambos, lo que explica la iniciación defectuosa del proceso de los rearreglos V(D)J. Abe y cols. en 1994 (32), reportaron defectos en la transcripción del gen Rag en células de la mo de otros pacientes con IDSC T-B-.
La IDSC ligada al cromosoma X, está caracterizada por un bloqueo en la maduración de los LT y de las células NK, con número normal o incrementado de LB. El locus de la IDSC ligada al X fue mapeado en la región Xq12. 13,1 (De Saint Basile y cols. 1987) (33).
Nogucchi y cols. en 1993 (34), reportaron que el gen que codificaba la cadena g (cg) del receptor de la IL-2, se localizaba en la misma región de la IDSC y en estos pacientes se encontraron mutaciones del gen cg.
La cg es común para los receptores de varias citoquinas: IL-2, IL4, IL-7, IL-9 e IL-15 y aumenta la afinidad por la respectiva citoquina y participa en la transducción de señales en los LT. El fenotipo de la IDSC ligada al X es una asociación compleja de defectos en estos cinco sistemas de citoquinas –receptor de citoquinas.
Las células T detectan la presencia del ag a través del heterodímero de superficie denominado:
Receptor para las células T (TCR). Estas moléculas no se expresan solas, ya que requieren la asociación de un grupo de proteínas monomórficas llamadas CD3 (CD3 g, d, e, z), que al parecer favorecen la expresión del TRC/CD3 y participan en la liberación de señales que conducen a la maduración de los LT o a la apoptosis en el timo, y a la activación de los LT o a la energía en la periferia (Alarcon y cols. 1993) (35).
Regueiro y cols. en 1986 (36), describieron la deficiencia de CD3 en humanos. Seis años más tarde, Arnaiz – Villena y cols. en 1992 (37), encontraron una deficiencia de CD3g. En 1993, Soudais y cols. (38), reportaron el defecto de CD3e. Miyasaki y cols. en 1994 (39), descubrieron que los receptores de las citoquinas que emplean la cg común, siempre se asocian con la tirosina kinasa intracelular JAK3. Éste y otros hallazgos proporcionaron indicios sobre las bases moleculares de la IDSC autosómica recesiva T-B+ NK. Macchi y cols. en 1995 (40), encontraron una reducción marcada en la expresión de la proteína JAK3 en estos pacientes debida a mutaciones en el gen JAK3 kinasa. Estos descubrimientos evidenciaron que la señal cg es mediada por la activación de JAK3.
Recientemente se han descrito IDP por alteraciones funcionales los LT, asociados con la producción anormal de citoquinas. Disanto y cols. en 1990 (41), hallaron un defecto en la síntesis de IL-2 en un paciente con IDSC; pero se han descrito otros pacientes con alteraciones múltiples en la generación de IL-4, IL-5 e IFNg (Weinberg y cols. 1990) (42).
Son múltiples los trastornos funcionales de los linfocitos T que se han descrito.
Partisetti y cols. en 1994 (43), verificaron una deficiencia en el flujo de Ca++ transmembrana. El defecto de la tirosina kinasa ZAP70 en un grupo de siete pacientes en cuatro familias, evidenció el papel que tiene la ZAP70 en la activación de los LT (Elder y cols. 1994) (44). En 1995, Fisher y cols. (45), definieron una enfermedad denominada síndrome linfoproliferativo autoinmune, el cual presenta un incremento en los LT CD3+ CD4-CD8-CD45RA+ y es causada por mutaciones en el gen Fas.
La Enfermedad Granulomatosa Crónica (EGC), ha sido un modelo fundamental para la investigación de la composición y activación del sistema microbicida de las células fagocíticas, en especial la de los neutrófilos. Esta entidad es causada por un defecto profundo en la explosión respiratoria que acompaña a la fagocitosis de todas las células mieloides (poly, eosinófilos, monocitos, mj).
La explosión respiratoria genera la conversión catalítica del oxígeno molecular al anión superóxido el cual se convierte en peróxido de hidrógeno, ácido hipocloroso y radicales hidroxilo. Estos derivados del oxígeno, realizan un papel importante en la reacción microbicida contra bacterias y hongos.
La enzima que cataliza la explosión respiratoria es la NADPH oxidasa (Curnutte y cols. 1975) (46), Ross y cols. 1999 (47). Se compone por lo menos de cuatro proteínas: gp91phox (phox: oxidasa de los fagocitos p22phox (las dos subunidades del citocromo b 558 que se encuentran en la membrana plasmática, centro redox de la oxidasa; descubierta por Segal y cols. en 1978 (48). Dos componentes citosólicos p47 phox y p67 phox. Estudios realizados con sistemas libres de células (Curnutte y cols. 1985 ) (49), revelaron que tanto las fracciones de la membrana plasmática, como las del citosol, se requieren para la activación de la oxidasa. Figura 1.
La EGC es causada por un defecto en cualquiera de estos cuatro componentes.
Como resultado del defecto para activar la explosión respiratoria de los fagocitos, la mayoría de las pacientes con EGC sufren infecciones recurrentes y severas: neumonías, linfadenitis, abscesos cutáneos y hepáticos, osteomielitis y septicemias; las cuales se inician durante el primer año de vida y son causadas especialmente por Staphylococcus aureus, Aspergillus spp, bacterias entéricas gram negativas. Además presentan granulomas difusos en diferentes sitios, que pueden ser lo suficientemente grandes para desarrollar síntomas dolorosos y obstructivos.

Figura 1. Sistema NADPH oxidasa de los Fagocitos. Activación: Los componentes gp91 phox, p22 phox y rap1, están localizados en las membranas de los gránulos específicos y las vesículas secretoras de los poly en reposo. Durante la activación celular, estas organelas se fusionan con la membrana plasmática, lo que permite la expresión de estas proteínas sobre la superficie de los poly. Simultáneamente los componentes citosólicos de este sistema transportador de electrones: p40 phox, p47 phox y p67 phox se trasladan a la membrana plasmática y se unen al citocromo b558 (subunidades gp91 phox y p22 phox), e inducen un cambio conformacional en la gp91 phox, permitiendo que la NADPH se una a esta glicoproteína y done sus electrones a la fracción FAD en esta subunidad.
La proteína rap1A puede modular negativamente la actividad de la oxidasa y las proteínas rac1 y rac2, intervendrían en la activación del sistema, al unirse a la p67 phox y al citocromo b558 respectivamente.
Las mutaciones en el gen que codifica la gp91 phox causa la EGC recesiva ligada al cromosoma X (65% de los pacientes). En el mundo se han encontrado diversos tipos de mutaciones que causan la EGC.
Nuestro grupo “Inmunodeficiencias Primarias” de la Universidad de Antioquia, hemos caracterizado los defectos genéticos y moleculares de dos proteínas (gp91 phox, p67 phox), que componen la NADPH oxidasa de los fagocitos responsables de la presentación de EGC.
El estudio de las mutaciones del gen CYBB (gp91 phox), en un paciente se encontró una sustitución de adenina por guanina en el penúltimo nucleótido del intrón 12, lo cual altera el sitio consenso de reconocimiento para el procesamiento del RNAm y por tanto conduce a la eliminación del exón 13 del RNAm maduro.
En la segunda familia analizada se demostró una sustitución de la citosina 271 en el exón 4 por timina, mutación sin sentido, la cual predice un cambio de Arg91 por un codón de paro prematuro.
El tercer paciente presentó una transversión de adenina:
Por timina en el último nucleótido del exón 6 lo cual predice la sustitución de Glu225 por Val. En otros dos pacientes se identificaron mutaciones sin sentido en el exón 5; uno presentó la sustitución de la guanina 318 por adenina, lo cual cambia Trp106 a un codón de paro, mientras el otro reveló un cambio de citosina en la posición 469 por timina que conduce al cambio de Arg157 por codón de paro. Un sexto paciente presentó un cambio de guanina por adenina en la posición 731 del exón 7, lo que predice un cambio de Cys244 por Tyr.
En el último paciente se identificó la sustitución de citosina por timina en la posición 868 del exón 8, lo cual genera un codón de paro prematuro en la Arg290.
El análisis de un grupo de pacientes con EGC producida por defectos de la p67 phox demostró en una paciente un cambio homocigoto de citosina por timina en el exón 4, el cual produce un cambio de la Arg102 por un codón de paro prematuro.
Dos hermanas presentaron una mutación homocigótica que consistió en una delección de 5 nucleótidos en el extremo 3’ del exón 13, lo cual cambia el marco de lectura y genera un codón de paro prematuro en el aminoácido 399. En otro paciente de una familia no relacionada se encontró esta misma delección homocigótica en el exón 13.
Otra paciente demostró en el DNA genómico el cambio homocigótico guanina por adenina en el primer nucleótido del intrón 4, lo cual conduce a un defecto severo en el procesamiento del RNA mensajero con pérdida de varios exones.
Otra paciente, reveló en uno de los alelos una delección de 9 nucleótidos en el exón 2:
La cual predice la eliminación de Lys19, Lys20 y Asp21 respetando el marco de lectura, mientras que en el otro alelo se identificó la misma mutación en el intrón 4 de la paciente precedente.
Finalmente, en la última familia estudiada se encontró que dos hermanas eran heterocigotas para la mutación del intrón 4 ya descrita en los dos casos precedentes y además presentaban, en el exón nueve, la delección de la adenina 728, lo cual predice un cambio del marco de lectura del RNAm y un codón de paro prematuro en la posición 270. (Patiño PJ y cols. 1999) (50), Patiño PJ y cols. 1999 (51).
Como se puede deducir de estos resultados, la EGC presenta una heterogeneidad muy grande en sus defectos genéticos moleculares, lo que en ciertas ocasiones se puede traducir en variabilidad de su presentación clínica. La mayoría de los pacientes tienen mutaciones que son específicas a sus familias.
La caracterización de los genes responsables de la EGC ha posibilitado un diagnóstico prenatal, empleando tejido de biopsias de vellosidades coriónicas (Hopkins y cols. 1992) (52).
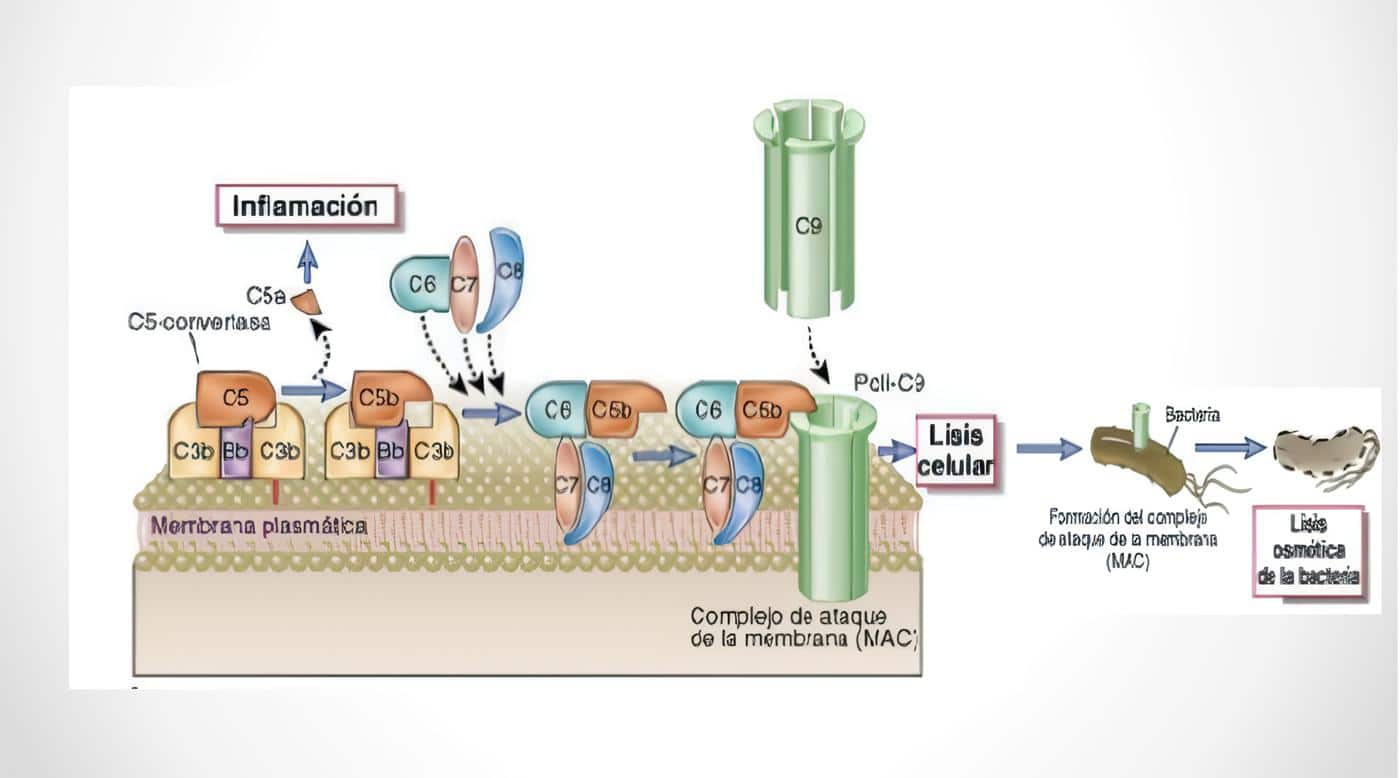
La complejidad de las IDP refleja la composición del SI. Bajo condiciones normales, la coestimulación de los linfocitos B por ags y las células T activadas por ags y citoquinas, lleva a la producción de acs, cuyos isotipos evolucionan de la IgM a la IgG y con una afinidad que se incrementa progresivamente; lo que da lugar a la formación de complejos ag-acs, los cuales son utilizados en la opsonización de microorganismos, en la neutralización de virus y en la activación de la cascada del complemento (C). Las subunidades del C que resultan de su activación, son agentes quimiotácticos para poly y mj.
Para emigrar de los espacios vasculares:
Las células fagocíticas necesitan citoesqueleto, moléculas de adhesión y la presencia de gradientes de factores quimiotácticos, lo que en conjunto permite su desplazamiento. Las células presentadoras de ag (CPA), están estratégicamente distribuidas en todo el organismo: mj, células dendríticas foliculares y células B. Para atrapar ag y presentarlos a las células T, los mj proveen varios tipos de receptores: Fc para las Igs y CR para el C.
En la Figura 2 se presenta un diagrama simplificado de los componentes mas importantes del SI. En él se resaltan algunos productos de los genes que son críticos para el desarrollo y la regulación de las respuestas inmunes y que están asociados con IDP, como se ha mencionado.

Figura 2. los genes que son críticos para Diagrama simplificado de los componentes más importantes del SI, y los productos de el desarrollo y regulación de las respuestas inmunes. Cada una de las moléculas encerradas por círculos, se han asociado con IDP hereditarias.
Las IDP monogénicas más frecuente observadas, son las ligadas al cromosoma X, y sólo necesitan la presentación de un evento mutacional único para que se manifiesten (Puck JM 1999) (53). Tabla 1.
En el desarrollo de las IDP autosómicas recesivas, se pierde la función de ambas copias de un gen en los cromosomas homólogos, por lo cual se espera que la ocurrencia de estas enfermedades sea más baja.
Con el análisis de las presentaciones clínicas y de los hallazgos de laboratorio, se ha logrado agrupar a las IDP humanas en una serie de síndromes en los cuales es posible reconocer disfunciones en cada uno de los componentes del SI. Hasta el presente se han definido unas cien enfermedades por deficiencia inmunológica. La clasificación de estas patologías se realiza periódicamente desde hace varios años, por un grupo de investigadores de la Organización Mundial de la Salud (WHO Scientific Group, 1997 (54).
Tabla 1. Genes de inmunodeficiencias ligadas al cromosoma X
Inmunodeficiencia |
Nombre
|
Proteína |
Localización |
Inactivación del Cromosoma X |
| Enfermedad Granulomatosa Crónica LX |
CYBB | Citocromo Oxidasa gp 91 phox |
Xp 21,1 | No |
| Síndrome Wiskott Aldrich |
WAS | Proteína WAS |
Xp 11, 22 | Si: Todas las células derivadas (mo) |
| Deficiencia properdina |
PFC | Properdina | Xp 11, 23 | No |
| IDSC LX | RG IL-2 | Cadena y común Receptores Citoquinas |
Xq 13,1 | Si: T, B, Nk |
| ALX | BTK | Tirosina Kinasa LB |
Xq 21, 3-22 | Si: LB |
| Síndrome Hiper IgM LX |
CD4OL | Ligando CD 40 |
Xq 26, 3-Q 27, 1 |
No |
| Síndrome Linfoproliferativo LX |
LYP (DSHP) |
DSP | Xq 25 | No |
| Deficiencia G6PD | G6PD | G6PD | Xq 28 | No |
Con una rata de incidencia estimada en 1 x 10.000 nacidos vivos y con una fuerte utilización de los servicios médicos en su tratamiento, el cuidado de los pacientes con IDP, crean un impacto en los servicios de atención en salud. Debido al gran número de IDP y a su amplia variedad de anormalidades, el estudio tamizado de la población no se ha considerado una opción práctica. Por tanto, es preciso fomentar un índice alto de sospecha a través de la educación de médicos y del público en general, para llegar a realizar oportunamente los diagnósticos.
El diagnóstico genético preciso transfiere grandes beneficios a los familiares de los pacientes y al sistema de atención en salud; porque aun pacientes con IDP ligadas al X, a menudo tienen una historia familiar negativa, y el reconocimiento de que la condición es genética es un avance importante. La predicción de la recurrencia de los riesgos, el consejo genético, pruebas prenatales y a los portadores, están disponibles para un gran número de IDP.
Se ponen de relieve algunas pruebas diagnósticas prenatales en ciertas IDP; en la deficiencia de la enzima adenosina deaminasa (ADA), el ensayo de ésta, en las vellosidades coriónicas, células amnióticas y sangre fetal (Hirschorn y cols. 1992) (55). Detección del síndrome de DiGeorge, IDSC debida a defectos de la tirosina kinasa JAK3 y otras más.
La detección de portadores de la EGC:
Se realiza investigando el patrón mosaico de oxidasa positiva y oxidasa negativa en los neutrófilos en una prueba de NBT, en la detección de la producción de peróxido de hidrógeno por citometría de flujo. Cuando se conoce una mutación específica en una familia, es más confiable realizar la detección de los portadores a nivel del DNA, empleando el ensayo SSCP (polimorfismos conformacionales de cadena de DNA sencilla), el cual puede ser informativo si se presenta un patrón anormal de la migración en una banda de DNA (García de OD y cols. 1997) (56), Patiño PJ y cols. 1999 (50), Patiño PJ y cols. 1999 (51).
Como se colige de esta exposición, la clasificación de estas enfermedades moleculares se está integrando con la contribución de ensayos in vitro aun más sofisticados para investigar el funcionamiento del SI. Este nivel nuevo de definición, sirve para perfeccionar las clasificaciones clínicas y de laboratorio establecidas, lo que hace posible un seguimiento clínico longitudinal y un ajuste en los tratamientos para cada genotipo.
La extensión lógica de este tipo de clasificación, es la recolección de la información de un gran número de pacientes con desórdenes genéticos particulares. Estas bases de datos contienen: presentación clínica, datos moleculares, incluyendo las mutaciones de genes específicos, los tratamientos y los seguimientos por períodos largos de tiempo y los desenlaces en cada caso. Las bases de datos proporcionarán una habilidad sin precedentes en el análisis de las IDP.
La primera base de datos con las primeras mutaciones de las IDP:
la base BTK, se inició en 1994 por Vihinem y cols. 1995 (57). Los laboratorios que investigan pacientes con ALX fueron invitados a participar en un grupo de estudio cooperativo internacional, el cual permanece abierto para todos los investigadores que deseen participar. Posteriormente, se han creado bases de datos similares para otras IDP definidas. (Vihinem y cols. 1999) (58). Tabla 2.
La información concerniente a los pacientes con IDP se ha colectado en un registro realizado por la Sociedad Europea para las IDP (ESID) (Abedi y cols. 1995) (59).
| BTKbase: Drs.Mauno Vihinen, Institute of Medical Technology, University of Tampere. Finland and C.I. Edvard Smith, Karolinska Institute, Huddinge, Sweden. |
| CD40Lbase and JAK3base: DR. Luigi Notarangelo, Department of Pediatrics, University of Brescia, Italy. |
| FASbase: Dr. Françoise Le Deist, Hôpital Necker Enfants Malades, Paris, France. |
| IFNgRIbase: Dr. Jean Laurent Casanova, Hôpital Necker Enfants Malades, Paris, France. |
| IL2RGbase: Dr. Jennifer M. Puck, Laboratory for Gene Transfer, NCHGR/NIH, Bethesda MD, USA |
| LAD-I and II: Dr. Amos Etzioni, Rambam Medical Center, Haifa, Israel. |
| WASPbase: Dr. Klaus Schwarz, University of Ulm, Germany. |
| X-CGDbase: Dr. Dirk Roos, Central Laboratory of the Netherlands Red Cross, Amsterdam, The Netherlands. |
En Latinoamérica, el LAGID (Grupo Latinoamericano para las IDP), constituido en 1993 por doce países, tiene como objetivo estudiar la frecuencia de las IDP en varias regiones del continente americano y aumentar el conocimiento sobre estas enfermedades.
Para este propósito, fue muy importante desarrollar un registro de las IDP, empleando un cuestionario uniforme y una base de datos computarizada.
En su primer informe (Zelasko y cols. 1998) (60):
Sobre 1428 pacientes, se reportó que las deficiencias de acs fueron los predominantes, 58%, seguidas por las IDP celulares y de acs asociadas con otras anormalidades, 18%, síndromes de IDP asociadas con disfunción de los granulocitos, 8%, trastornos fagocíticos, 9%, ID celular y de acs, 5%, y defectos del C, 2%.
En Antioquia, nuestro grupo “Inmunodeficiencias Primarias” de la Universidad de Antioquia, a partir de agosto de 1994 hasta diciembre de 1999, ha caracterizado 65 casos de IDP. (Clasificación IDP: LAGID 1997 (61), reporte del Comité Científico de la IUIS 1999 (62). Tablas 3, 4.
El registro realiza una lista de diferentes desórdenes de IDP, diversos valores de investigaciones en el laboratorio, historia familiar y genética de los pacientes y terapia. Vihinem y cols. 1999 (58), le sugieren a los médicos, enviar la información de sus pacientes a los sitios de la red mundial por Internet (WWW). Tabla 5.
Tabla 3. Clasificación fenotípica: 65 Inmunodeficiencias Primarias. Agosto 1994 – Diciembre 1999. Grupo de Inmunodefiencias Primarias. Universidad de Antioquia. Medellín
DEFICIENCIAS PREDOMINANTES DE ANTICUERPOS (30) |
|
| · Hipogammaglobulinemia Transitoria de la infancia | 10 |
| – Inmunodeficiencia común variable | 6 |
| – Agammaglobulinemia ligada al X | 6 |
| – Deficiencias de anticuerpos con inmunoglobulinas normales | 3 |
| · Deficiencia selectiva de IgA | 2 |
| · Inmunodeficiencia común variable con timoma | 1 |
| · Síndrome de Hiper IgM | 1 |
| · Deficiencia selectiva de IgG2 | 1 |
INMUNODEFICIENCIAS ASOCIADAS A DEFECTOS
|
|
| · Síndrome de Hiper IgE con Infecciones Recurrentes | 11 |
| · Síndrome de Chediak-Higashi | 1 |
DEFICIENCIAS CELULARES Y DE ANTICUERPOS
|
|
| · Síndrome de Wiskott Aldrich | 4 |
| · Ataxia Telangiectasia 2 · Candidiasis Mucocutánea Crónica | 2 |
DEFICIENCIAS COMBINADAS (8) |
|
| · Inmunodeficiencia severa combinada | 5 |
| · Inmunodeficiencia celular con Igs normales | 2 |
| · Inmunodeficiencia combinada no severa | 1 |
DEFECTOS PRIMARIOS DE LAS CÉLULAS FAGOCÍTICAS (6) |
|
| · Deficiencia del Receptor IFN g | 3 |
| · Síndrome de Enfermedad Granulomatosa Crónica | 2 |
| · Enfermedad de Kostmann | 1 |
DEFICIENCIAS DEL COMPLEMENTO (1) |
|
| · Edema angioneurótico | 1 |
Comunicación personal: Dr. Carlos Montoya G. Coordinador. Programa: Síndrome Infección Recurrente. Grupo inmunodeficiencias Primarias. Universidad de Antioquia.
Tabla 4. Inmunodeficiencias Primarias. Programa SIR – IDP Universidad de Antioquia
| Deficiencia predominante de anticuerpos | 30 | 47% |
| IDP Asociadas a defectos de células Fagocíticas | 12 | 19 % |
| Deficiencias combinadas | 8 | 12 % |
| Deficiencias celulares y de anticuerpos asociadas con defectos mayores |
8 | 12 % |
| Defectos células fagocíticas | 6 | 9 % |
| Defectos complemento | 1 | 1 % |
Comunicación Personal: Dr. Carlos Montoya G. Coordinador. Programa: Síndrome Infección Recurrente (SIR). Grupo Inmunodeficiencias Primarias. Universidad de Antioquia
La correlación entre la enfermedad clínica por IDP y el fenotipo, ha generado una nueva fase en el desarrollo del diagnóstico genético específico y en el manejo médico ajustado al genotipo del paciente. Así, la IDSC debida a la deficiencia de ADA, se puede tratar con reemplazo de la enzima ADA conjugada con polietilenglicol, en forma alternativa, la terapia con trasplante de mj proveniente de un donante histocompatible o con mj depletada de células T haploidénticas.
Se están llevando a efecto otros manejos prometedores de las IDP: terapia con genes, la cual se inició con la transducción de un gen ADA normal ex vivo; la amplificación de las células T del paciente ex vivo y el trasplante con sangre de cordón autólogo; ésta última en niños diagnosticados en la etapa prenatal (Stiehm ER, 1999) (63).
Estos análisis de los pacientes con IDP, nos ayudan a comprender más profundamente como el hombre puede vivir libre de infecciones, rodeado de un universo pleno de microorganismos.
Finalmente, los descubrimientos que se han descrito nos indican en que lugar estamos con relación al conocimiento de las IDP, y nos señalan la ruta por la cual debemos dirigirnos para proseguir el análisis de los fenómenos por deficiencias del sistema inmune.
Referencias Bibliográficas
- 1. Bruton OC. Agammaglobulinemia. Pediatrics. 1952; 9:722-727.
- 2. Janeway CA, Apt L, Gitlin D. Agammaglobulinemia. Trans Assoc Am Physicians 1953; 66:200-202.
- 3. Good RA, Zak SJ. Disturbances in gamma globulin synthesis as “Experiments of Nature”. Pediatrics 1956; 18:109-149.
- 4. Good RA, Kelly WD, Rötstein J, Varco RL. Immunological deficiency disease and Sarcoidosis. Prog Allergy. 1962; 6: 187-319.
- 5. Cooper MD, Faulk PW, Fudenberg HH, et al. Classification of Primary immunodeficiencies. N Eng J Med. 1973;288: 966-967.
- 6. Good RA. Progress Toward a cellular engineering. Jama 1970; 214:1289.
- 7. Kunkel HG, Slater R, Good RA. Relation between certain myeloma proteins and normal gamma globulin. Proc Soc Exp Biol Med. 1995; 76:190-193.
- 8. Good RA, Agammaglobulinemia -a provocative experiment of nature. Bull Univ Minn Med Found. 1954; 26:1-19.
- 9. Good RA, Rotstein J, Mazzitello WF. The simultaneous ocurrence of rheumatoid arthritis and agammaglobulinemia. J Lab Clin Med. 1957; 39:343-357.
- 10. Good RA, Junis EJ. Association of autoimmunity, immunodeficiency and aging in man, rabbits and mice . Fed Proc. 1974; 33:2040-2050.
- 11. Good RA, Varco RL. A Clinical and experimental study agammaglobulinemia. Lancet 1955; 75: 245.
- 12. Cooper MD, Peterson RDA, South AM, Good RA. The functions of the thymus system and the bursa system in the chicken. J Exp Med. 1996; 123: 75.
- 13. Cooper MD, Cain WA, Van Alten PJ, Good RA. Development and function of the immunoglobulin – producing system: I Effect of bursectomy at different stages of development on germinal centers plasma cells, immunoglobulin and antibody production. Int Arch Allergy Appl Immunol. 1969; 35:242.
- 14. Lodbell DH. Congenital absence of the parathyroid gland. Arch Pathlol. 1959; 67: 412-418.
- 15. DiGeorge AM. Discussions on a new concept of the cellular basis of Immunology. J Pediatr. 1965; 67: 907.
- 16. Conley ME, Becwith JB, Mancer JFK, Tenckhoff L. The spectrum of the DiGeorge Syndrome. J Pediatr 1979; 94:883-890.
- 17. Gatti RA, Meuwissen HJ, Allen HD, et al. Immunological reconstitution of Sex-Linked Lymphopenic Immunological Deficiency . Lancet. 1968; 2: 1366-1369.
- 18. Cleveland WW, Fogel BJ, Brown WT, Kay HEM. Foetal Thymic Transplant in a case of DiGeorge Syndrome. Lancet. 1968; 2:1211-1214.
- 19. Cooper MD, Lawton AR, Bockmann DE. Agammaglobulinemia with B Lymphocytes: specific defect of plasma cell differentiation. Lancet 1971; 2: 791.
- 20. Seigal FP, Pernis B, Kunkel HG. Lymphocytes in Human immunodeficiency states: a study of membrane-associated immunoglobulin. Eur J Immunol. 1971; 1: 482.
- 21. WU LFY, Lawton AR, Cooper MD. Differentation capacity of cultured B Lymphocytes from immunodeficient patients. J Clin Invest. 1973;52:3180.
- 22. Pearl ER, Vogler LB, Okos AJ. Lymphocytes precursors in human bone marrow: an analysis of normal individuals and patients with antibody deficiency states. J Immunol. 1978; 120:1169.
- 23. Naor D, Bentwich Z, Cividalli G. Inability of peripheral lymphoid cells of agammaglobulinemic patients to bind radioiodinated albumins. Aust J Exp Biol Med Sci. 1969; 47: 759-761.
- 24. Sideras P, Smith CIE. Molecular and cellular aspects of X-Linked agammaglobulinemia. Adv Immunol. 1995; 59: 135-223.
- 25. Royer – Pokora B, Kunkel LM, Monaco AP, et al Cloning the gene for an inherited human disorder – Chronic Granulomatous Disease – on the basis of its chromosomal location. Nature 1986; 322: 32-38.
- 26. Vetrie D, Vorechovsky I, Sideras P, et al. The gene involved in X-Linked agammaglobulinemia is a member of the Src family of protein – tyrosine kinases . Nature. 1993; 361: 226-233.
- 27. Tsukada S, Saffran DC, Rawlings DJ, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993; 72: 279-290.
- 28. Graf, Korthäer U, Mages HW, Senger G, Kroczek RA. Cloning of trap, a ligand for CD40 on human T cells. Eur J Inmunol. 1992; 22: 3191-3194.
- 29. Schwarz K, Hansen – Hagge TE, Knoblock C, et al. Severe combined immunodeficiency (SCID) in man: B cell – negative (B-) SCID patients exhibit an irregular recombination pattern at the JH Locus. J Exp Med. 1991; 174: 1039-1048.
- 30. Cavazzana – Calvo M, Le Deist F, De Saint Basile G, Paradopoulo D. Increased radiosensitivity of granulocyte – macrophage colony forming units and skin fibrobrast in human autosomal recessive severe combined immunodeficiency. J Clin Invest. 1993; 91: 1214-1219.
- 31. Schwarz K, Gauss GH, Ludwing L, et al. Rag mutations in human B cell- negative SCID. Science. 1996; 247: 97-99.
- 32. Abe T, Tsuge I, Kamachi Y, et al. Evidence for defects in V(D)J rearrangements in patients with severe combined immunodeficiency. J Immunol. 1994; 152:5504-5513.
- 33. De Saint Basile G, Arveiler R, Oberlé I, et al. Close linkage of the locus of X chromosome linked severe combined immunodeficiency to polymorphic DNA markers in Xq 11-13. Proc Natl Acad SCI USA. 1987; 84:7576-7583.
- 34. Nogucchi M, Yi H, Rosenblatt HM, et al. Interleukin-2 receptor g chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993; 3:147-156.
- 35. Alarcon B, Terhorst C, Timón M, et al. Congenital T-cell Receptor Immunodeficiencies in Man. In Immunodeficiencies. Rosen FS, Seligmann M, eds. Harwood Academic: Chur (Switzerhland). 1993, 155-166.
- 36. Regueiro JR, Arnaiz – Villena A, Ortiz De Landazuri M, et al Familial defect of CD3 (T3) expression by cells associated with rare gut epithelial cell autoantibodies. Lancet. 1986; i: 1274-1275.
- 37. Arnaiz-Villena A, Timón M, Corell A, et al. Brief report: primary immunodeficiencies caused by mutations in the gene encoding the DC3g subunit of the T-lymphocyte receptor. N Engl J Med 1992; 327: 529-533.
- 38. Soudais C, Villarty JP, Deist Fl, et al. Independent mutations on the human CD3e gene resulting in a T cell receptor/CD3 immunodeficiency. Nat Genet. 1993; 3: 77-81.
- 39. Miyazaki T, Kawahara A, Fujii H, et al. Functional activation of JAK1 and JAK3 by selective association with IL-2 receptor subunits. Science. 1994; 266: 1045-1047.
- 40. Macchi P, Villa A, Giliani S, et al. Mutations of JAK3 gene in patients with autosomal severe combined immunodeficiency (SCID). Nature 1995; 377: 65-68.
- 41. Disanto JP, Keever CA, Small TN, et al. Absence of interleukin-2 production in a severe combined immunodeficiency disease syndrome with T cells. J Expl Med. 1990;171: 1697-1705.
- 42. Weinberg K, Parkman R. Severe Combined immunodeficiency due to a specific defect in the production of IL-2. N Engl J Med. 1990; 322: 1718-1725.
- 43. Partisetti M, Le Deist F, Hivroz C, et al. The calcium current activated by T-cell receptor and store depletion in humans lymphocytes is absent in a primary immunodeficiency. J Biol chem 1994; 269:32327-32335.
- 44. Elder ME, Lin D, clever J, et al. Human SCID due to a defect in Zap70, a T cell tyrosine kinase. Science. 1994; 264:4596-4599.
- 45. Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering fas gene mutation impair apoptosis in a human autoinmune lymphoproliferative syndrome. Cell. 1995; 81: 935-946.
- 46. Curnutte JT, Whitten DM, Babior BH. Defect in pyridine nucleotide dependent superoxide production by a particulate fraction from the granulocytes of patients with chronic granulomatous disease. N Engl J Med 1975; 293: 628-632.
- 47. Ross D, Curnutte JT, Chronic Granulomatous Disease. In Ochs HD, Smith CIE, Puck JM, eds. Primary Immunodeficiency Diseases. A molecular and Genetic approach. New York: Oxford University press, 1999;353-374.
- 48. Segal AW, Jones OTG, Webster D, Allison AC. Absence of a newly described cytochrome b from neutrophils of patients with Chronic Granulomatous Disease. Lancet. 1978; 2: 446-449.
- 49. Curnutte JT. Activation of human neutrophil nicotinamide adenine dinucleotide phosphate, reduced/triphosphopyridine by arachidonic acid in a cell-free system. J Clin Invest. 1985; 75:1740-1743.
- 50. Patiño PJ, Pérez JE, López JA, et al. Molecular Analysis of Chronic granulomatous disease caused by defects in gp 91-phox. Hum mutat 1999; 13:29-37.
- 51. Patiño PJ, Rae J, Noack D, et al. Molecular characterization of autosomal Recessive Chronic Granulomatous Disease Caused by a defect of the Nicotinamide Adenine Dinucleotide Phosphate (Reduced Form) Oxidase Component p67 phox. Blood 1999; 94:2505-2514.
- 52. Hopkins PJ, Bemiller LS, Curnutte JT. Chronic granulomatous disease: diagnostic and classification at the molecular level. Clin Lab Med. 1992; 12: 277-304.
- 53. Puck JM. Genetic aspects of Primary Immunodeficiencies. In Ochs HD, Smith CIE, Puck JM, eds. Primary Immunodeficiency Diseases.
- 54. WHO Scientific Group. Primary Immunodeficiency Disease. Clin Exp Immunol 1997; 109 (suppl) 1: 1-28. deaminase deficiency and selected other immunodeficiencies. In Milunski A, ed. Genetic Disorders and the Fetus: Diagnosis, Prevention and Treatment. 3ed. New York: Plenum Press. 1992; 453-464.
- 55. Hirschorn R. Prenatal diagnosis of adenosine A Molecular and Genetic Approach. New York: University Press. 1999; 432- 442.
- 56. García de OD, Patiño PJ, Salgado H, et al. Evaluación del Paciente con Inmunodeficiencia. Síndrome de Infección Recurrente Patológica. Medicina & Laboratorio. 1997;7: 545-575.
- 57. Vihinem M, Cooper MD, de Saint Basilé G, et al. BTK base: a database of XLA causing mutations. Immunol Today. 1995;16: 460-465.
- 58. Vihinem M, Lehväslaiho H, Cotton RGH. Immunodeficiency Mutation Databases. In Ochs HD, Smith– CIE, Puck JM, eds. Primary Immunodeficiency Diseases. A Molecular and Genetic Approach. New York: Oxford University Press. 1999;443-447.
- 59. Abedi MR, Morgan G, Paganelli R. Report from the EGID registry of primary immunodeficiencies. In Caracol I Espanol T, Fontan G, Matamoros N, eds. Progress in Immune Deficiency V Barcelona: Springer Verlag. 1995;113-115.
- 60. Zelasko M, Carneiro Sampaio M, Cornejo de Luigi M, García de OD, et al. Primary Immunodeficiency Diseases in Latin America. First Report from Eight Countries participating in the LAGID J Clin Immunol. 1998;18:161-166.
- 61. Clasificación Fenotípica de las Inmunodeficiencias Primarias. 1997; 1:11-18.
- 62. Primary Immunodeficiency Diseases. Report of an IUIS Scientific Committee. Clin Exp Immunol 1999; 118(suppl.1):1-28.
- 63. Stiehm ER. Conventional Therapy of Primary Immunodeficiency Diseases. In Ochs HD, Smith CIE, Puck JM, eds. Primary Inmmunodeficiency Diseases. A Molecular and Genetic Approach. New York: Oxford University Press. 1999; 448-458.