Fisiología de la Corteza Suprerrenal
Autor: Alfredo F. Jácome-Roca, MD
Generalidades – Fisiología de la Corteza Suprerrenal
Esta estructura de origen mesodérmico, que constituye el 90% de las glándulas suprarrenales, es la encargada de producir glucocorticoides y mineralocorticoides. Estos son esteroides del grupo C 21, y son el cortisol (o hidrocortisona) por un lado y la aldosterona por el otro. En el primer grupo también juegan un papel la corticosterona (principal hormona glucorreguladora en roedores) y la cortisona. En el segundo grupo está además la deoxicorticosterona o DOCA.
El cortisol se encarga –a la par de otras hormonas- de regular la homeostasis de la glucosa y el manejo del estrés. Eleva la glicemia, al igual que lo hacen la somatotropina, el glucagòn y la epinefrina. Hace parte pues de las hormonas contrarreguladoras de la insulina. Es también inmunosupresor (linfolìtico) y antinflamatorio, y juega un papel permisivo en la acción de otras hormonas.
Así mismo, la aldosterona por su parte regula la volemia por su capacidad retenedora de sodio. Haciendo parte de los mecanismos de regulación del metabolismo hídrico y electrolítico y de la tensión arterial.
También se producen en la corteza andrógenos de poca potencia, como la dehidroepiandrosterona sulfato (DHEA-S) y algunos estrógenos en pequeñas cantidades.
Anatomía e histología – Corteza Suprerrenal
Las glándulas suprarrenales son estructuras aplanadas en forma de gorro. Se encuentran localizadas sobre el polo superior de ambos riñones. A nivel de la primera vértebra lumbar. Pesan alrededor de 5 g cada una y están rodeadas por un colchón de tejido adiposo. El cual a su vez se haya cubierto por una delgada cápsula de tejido fibroso, adherida a la glándula por numerosas bandas. Este compartimiento se denomina Fascia de Gerota.
Por otro lado, aunque de pequeño tamaño, son esenciales para la vida. Básicamente por su producción de mineralocorticoides. La corteza constituye el 90% del volumen de la masa glandular. Y tiene un color amarillo dorado al corte, diferente del café oscuro de la medula.
Así mismo, aunque anatómicamente estas dos estructuras se encuentran en la misma localización. Se trata de dos órganos con su propia embriología. (La medula se origina de la cresta neural del ectodermo. Mientras que la corteza viene del mesodermo). Además son diferentes histológica y funcionalmente.
Por ende, la histología de la corteza muestra tres capas celulares epiteliales que por su contenido lipídico se les llama “células de jabón”. Están dispuestas en bandas irregulares alrededor de sinusoides.
La primera capa es delgada y se encuentra inmediatamente debajo de la cápsula e interviene primordialmente en la producción de mineralocorticoides. Se llama zona glomerulosa formada por células cuboidales en grupos ovoides, que recuerdan glomérulos. Depende del eje renina-angiotensina y es muy poco influida por la corticotropina o ACTH.
Además, la zona fasciculada es la más ancha, se encuentra debajo de la anterior, sus células están dispuestas en forma radiada, alineadas en cordones paralelos. La otra zona es la reticular, que tiene células acordonadas que forman una red alrededor de la medula.
Finalmente, estas dos últimas zonas están comprometidas en la esteroidogénesis de glucocorticoides, andrógenos, estrógenos y progestágenos. Y se encuentran bajo la influencia directa de la corticotropina (Figura 1).
 Figura 1. Histología de las cápsulas suprarrenales.
Figura 1. Histología de las cápsulas suprarrenales.
Regulación fisiológica de la corteza suprarrenal
La producción de esteroides de la corteza está básicamente controlada por el ACTH. Cuyas características se discutieron en el capítulo sobre adenohipófisis. Baste aquí decir que su secreción está básicamente regulada por la producción de cortisol (hidrocortisona) y de cortisona, en sus formas libres.
Además, los estrógenos y andrógenos no influyen para nada en su secreción. Y otros esteroides como la aldosterona, deoxicorticosterona y progesterona sólo lo hacen ligeramente. La corteza responde al ACTH con un mecanismo de “todo o nada” a una dosis de 0.35 unidades. Necesitándose una unidad para el funcionamiento cortical en 24 horas.
Así mismo, el cortisol efectúa una retroalimentación negativa sobre la hormona liberadora de corticotropina (CRH) producida en el hipotálamo, que frena la producción de ACTH. Lo que hace lo mismo con la producción de cortisol lo que a su vez favorece nuevos pulsos en el eje.
También, el cortisol es un esteroide C21. Que tiene dos hidroxilos en su cadena lateral que permiten una reacción calorimétrica que se usó mucho en el pasado. Por lo que se denominaba (al igual que otros corticoides de menor importancia), cromógeno de Porter Silber (Figura 2).
 Figura 2. Eje Hipotálamo-Hipófisis-Suprarrenales con feed-back negativo.
Figura 2. Eje Hipotálamo-Hipófisis-Suprarrenales con feed-back negativo.
 Figura 2. Eje Hipotálamo-Hipófisis-Suprarrenales con feed-back negativo.
Figura 2. Eje Hipotálamo-Hipófisis-Suprarrenales con feed-back negativo. Por ende, la producción de aldosterona depende del sistema renina-angiotensina. La renina es una enzima (aspartil proteasa, peso molecular 38 kDa). Generada en el aparato yuxtaglomerular del riñón. (Constituido el barostato por la parte distal de la arteriola aferente, la proximal de la eferente. Y el sodiostato y clorostato por las células yuxtaglomerulares. Situadas junto al primer segmento del túbulo contorneado distal que se denomina mácula densa). La renina se libera por cambios hemodinámicos en la arteriola eferente.
Igualmente, estos pueden ser la pérdida de sangre, cambios posturales o disminución en los niveles de sodio. Una globulina alfa 2 (de 60 kDa) originada en el hígado. El angiotensinógeno. Es catalizada por la renina para producir el decapéptido inactivo conocido como angiotensina I. Por medio de la enzima convertidora de angiotensina –mejor conocida como ECA-. Esta pierde dos aminoácidos y se transforma en el octapéptido angiotensina II.
Adicional a esto, esta última hormona interactúa con un receptor suprarrenal con el que tiene gran afinidad y produce la aldosterona (Figura 3). Es una hormona muy potente, que aunque circula sólo por un minuto antes de ser destruida en el pulmón. Tiene varios efectos, todos destinados a elevar la presión arterial: estimula el simpático a varios niveles e inhibe el tono vagal. Estimula la sed, el apetito por la sal. La hormona antidiurética, el ACTH y como ya dijimos, la producción de aldosterona.
Figura 3. Esteroidogénesis.
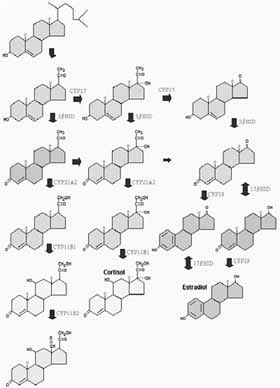 Figura 3. Esteroidogénesis.
Figura 3. Esteroidogénesis.Además, este último mineralocorticoide es un esteroide C21, que tiene un grupo aldehido (CHO) en el carbono 18. Y sólo cuenta con un hidroxilo en la cadena lateral. Características de estructura química que lo diferencian del cortisol.
Finalmente, el sistema renina-angiotensina actúa de manera antagónica al Péptido Auricular Natriurético. Que además de la acción que le da su nombre es relajante del músculo liso vascular. Inhibe la vasoconstricción por la norepinefrina y la angiotensina II, reduce la secreción de renina y de aldosterona.
Esteroidogénesis
Las hormonas esteroides son derivadas del colesterol. El que proviene del sintetizado en la célula a partir del acetato, o de los depósitos de ésteres de colesterol. Que se encuentran en las vacuolas lipídicas intracelulares o a partir de la captación de lipoproteínas de baja densidad, que son ricas en colesterol. Esta última fuente es la más importante cuando una estimulación crónica de las células capaces que realizar la esteroidogénesis.
La reacción inicial de la esteroidogénesis (conversión de colesterol a pregnenolona) se observa en todas ellas. Es decir, en las gónadas (ovarios y testículos), suprarrenales, placenta y algunas células del sistema nervioso central. Los pasos que siguen –con algunas excepciones- son comunes tanto a la corteza suprarrenal como a las gónadas.
Además, la principal diferencia consiste en que la corteza contiene las hidroxilasas 21 y 11. Que permiten la formación de algunos corticosteroides del tipo C 21 (cortisol, aldosterona, corticosterona) que las gónadas no pueden producir. Aunque la corteza produce esteroides C 19 (andrógenos) y C18 (estrógenos) estos son de escasa importancia al relacionarlos con los gonadales.
Así mismo, la esteroidogénesis incluye la formación de más de 40 esteroides derivados del ciclopentano-perhidrofenantreno. Pero los que tienen mayor importancia fisiológica son sólo unos pocos. En todas estas transformaciones actúa el citocromo P 450. Y que la reacción inicial (colesterol a pregnenolona). Se realiza en la mitocondria gracias a la acción de una enzima de la membrana interna que se llama CYP11A1, reacción bastante limitada.
Luego, en los pasos posteriores juegan un papel activador importante las hormonas tróficas ACTH –corteza-. FSH. LH –gónadas-. Y la angiotensina II –capa glomerular.- Para dar como resultado el esteroide final. La batería de enzimas oxidativas localizadas en la mitocondria y en el retículo endoplásmico. Genera la biosíntesis esteroidea. Que se limita cuando se frena el transporte de colesterol libre citoplasmático a la mitocondria.
También, la formación de cortisol (Figura 1) se produce como sigue:
La pregnenolona se transforma en progesterona, por acción de la 3 beta-HSD. Esta, por acción de la CYP 17, en 17 hidroxi-progesterona. La acción de la CYP 21 A2 produce la 17 alfa- 21 dihidroxi- progesterona (11-deoxicortisol o compuesto S). Que se hidroxila a nivel del carbono 11 (CYP 11 B1). Para dar lugar al cortisol o hidrocortisona, que se puede intercambiar a nivel periférico con la cortisona.
Por ende, la tabla 1 muestra cuáles son las enzimas comprometidas en la esteroidogénesis. Y la figura 4 los diferentes esteroides generados en los tejidos con capacidad para hacerlo, particularmente corteza y gónadas.
También, la formación de aldosterona (Figura 1) se produce como sigue:
La progesterona (que proviene del colesterol y de la pregnenolona). Se transforma en desoxicorticosterona (DOCA) por acción de la CYP 21 A2. La DOCA en corticosterona (glucocorticoide) por medio de la enzima CYP 11 B1. Finalmente esta en aldosterona, por acción de la CYP 11 B2. La zona glomerulosa no expresa la CYP 17 y por esto no forma ni cortisol ni andrógenos.
Además, la dehidroepiandrosterona (DHEA), principal andrógeno suprarrenal, se produce (Figura 1) por la acción de la CYP 17 en dos oportunidades. La pregnenolona sufre inicialmente una 17-hidroxilación. Y luego por una degradación oxidativa de la cadena lateral 17-OH, 20-ceto, se produce la DHEA (que circula en su forma de sulfato (DHEA-S).
Finalmente, este compuesto es el principal 17- cetosteroide. Por lo cual la función androgénica suprarrenal podía medirse por esta determinación, la que la testosterona no es un 17-cetosteroide. Una dehidrogenización en el carbono 3 produce la androstenediona (Figura 1) que puede transformarse en testosterona (en las células de Leydig del testículo). Estos dos andrógenos, se aromatizan (por acción de aromatasas) para formar estrógenos del tipo estradiol o estrona.
Tabla 1
Enzimas de la esteroidogénesis
|
Nombre común |
Antiguo Nombre |
Actual Nombre |
|
Desmolasa, enzima de clivaje de la cadena lateral |
P450 SCC |
CYP11A1 |
|
3-betahidroxiesteroide- dehidrogenasa |
3 beta-HSD |
3 beta-HSD |
|
17 alfa hidroxilasa/ 17,20 liasa |
P450 C17 |
CYP 17 |
|
21-hidroxilasa |
P450 C21 |
CYP 21 A2 |
|
11-beta hidroxilasa |
P450 C11 |
CYP 11 B1 |
|
Aldosterona sintasa |
P450 C11 AS |
CYP 11 B2 |
|
Aromatasa (transforma andrógenos en estrógenos) |
P450 aro |
CYP 19 |
Patología suprarrenal.
Enfermedad de Cushing
El hipercortisolismo se ha denominado Enfermedad de Cushing. Cuando su causa es una hiperproducción de ACTH por parte de un pequeño adenoma central hipofisiario. Constituido por células anteriormente denominadas basófilas. Esta enfermedad constituye el 60% de los casos y tiene un tratamiento neuroquirúrgico.
Adenomas funcionantes
Los adenomas funcionantes suprarrenales unilaterales son otro 30%. Y requieren la extirpación de la glándula afectada, generalmente por lumbotomía. Los demás casos son carcinomas suprarrenales, que requieren tratamiento quimioterápico adicional. Y que se caracterizan –cuando son en mujeres- por marcada masculinización.
Producción ectópica de corticotropina
Existen los casos de producción ectópica de corticotropina, que frecuentemente se origina en cánceres pulmonares, y que cursan con marcada hipokalemia. Además de estos casos donde la aparición es espontánea, existen los más comunes que se observan en los pacientes bajo terapia crónica de corticoides.
Los casos de Cushing presentan pérdida de masa muscular, hipertensión, hiperandrogenismo. En mujeres (acné, hirsutismo, amenorrea). Distribución centrípeta de grasa corporal que incluye el “morro de búfalo”. Poliglobulia, hipokalemia, diabetes y con el tiempo, osteoporosis.
Enfermedad de Addison
La insuficiencia suprarrenal crónica se denomina Enfermedad de Addison. Y es causada más frecuentemente por problemas autoinmunes (donde puede hacer parte de un síndrome poliglandular autoinmune). O por infecciones del tipo tuberculosis.
Cuando se sospecha esta última patología, la terapia glucocorticoide (y mineralocorticoide) debe acompañarse de tratamiento antituberculoso. Los síntomas consisten en marcada debilidad, letargo, diarrea, hipotensión (que puede llevar a shock o a síncopes). Microcardia, hiperkalemia y pigmentación característica de la piel.
Hiperplasia suprarrenal
Otra patología suprarrenal que se observa particularmente en niños (en su variedad congénita) es la hiperplasia suprarrenal. La que es debida a diferentes deficiencias de las enzimas que intervienen en la esteroidogénesis. La deficiencia más común es la deficiencia parcial de CYP 21 A2. Que impide la 21 hidroxilación que da lugar al cortisol y acumula su precursor 17- hidroxi-progesterona. Compuesto que no suprime la secreción de ACTH, lo que lleva a estimulación crónica de la corteza y a hiperplasia suprarrenal. Con marcada producción de andrógenos.
Esto causa hipertrofia clitoridiana y progresiva virilización en niñas, y a macrogenitosomìa precoz en niños. La variedad que cursa con bloqueo enzimático completo produce una deficiencia mineralocorticoide que lleva a la muerte por deshidratación. Si no se detecta y corrige prontamente el problema. Otras deficiencias cursan con virilización e hipertensión. O pueden ser incompatibles con la vida, según el caso, y lo inicial del bloqueo en la síntesis de los corticoides. Como esta enfermedad es familiar, es posible detectarla (y tratarla) “in utero”.
Enfermedad de Conn
En cuanto a los mineralocorticoides, el síndrome menos raro es la Enfermedad de Conn. Donde hay hipertensión con hipocalemia. Y a veces, parálisis muscular. Hay una nueva serie de síndromes clínicos como el de Exceso Aparente de Mineralocorticoides (AME). Causado por una mutación del gen 17-B-HSD-tipo 2.
Pseudohiperaldosteronismo
El Pseudohiperaldosteronismo. (Incluye el Síndrome de Liddle y es autosómico recesivo. La hipertensión Juvenil, que cursa con hipokalemia, hipoaldosteronismo e hiporreninemia.
El Pseudohipoaldosteronismo tipo I causado por una mutación con pérdida de función del receptor mineralocorticoide. Finalmente una clase de hipertensión gravídica por sensibilidad a la progesterona. En la que existe una mutación del gen del receptor mineralocorticoide. Con sensibilidad a la sal y se da en homocigotos con alelos A7/A7.
Terapéutica.
Los corticoides pueden usarse como terapia de suplencia en la insuficiencia suprarrenal primaria y secundaria. Como terapia supresiva en la hiperplasia suprarrenal congénita. Por su efecto antialérgico en reacciones de este tipo. Por su efecto inmunosupresor en diferentes tipos de cáncer y otras patologías como el síndrome nefrótico. Y por su efecto antiinflamatorio en artritis. Y otras inflamaciones crónicas, de tipo sistémico o por acción local en piel y órganos de los sentidos.
También se utilizan en algunos casos de shock. Y para obtener la maduración pulmonar en neonatos pretérmino. Una patología muy común, hoy reconocida como inflamatoria, y que se beneficia enormemente de los corticoides. (Particularmente los inhalados). Es el asma bronquial. El mineralocorticoide 9-alfa-fluoro- hidrocortisona se usa en la suplencia de la Enfermedad de Addison.
Nota histórica.
Eustaquio (1563, “Opúsculos Anatómicos”). Las llamó cápsulas atrabiliarias, órganos huecos llenos de sangre oscura. Y más tarde Jean Riolan (1629) las denominó cápsulas suprarrenales. La Academia de Ciencias de Burdeos intentó averiguar cuáles eran las funciones de estas glándulas por medio de un premio. Pero Montesquieu tuvo que declararlo desierto en 1718.
El siglo XIX mostró alguna luz al final del túnel. Ya que desde el punto de vista anatómico se describieron las zonas de la corteza: reticular y fascicular. (Ecker, 1846). Y glomerulosa (Arnold, 1866). En clínica, Addison describió en 1849 un caso de anemia con insuficiencia suprarrenal. Y en 1855 once pacientes más. Brown-Séquard en 1856 observó que la adrenalectomía experimental mata los animales de experimentación. Tigerstedt y Bergman en 1898 descubrieron la renina.
El siglo XX tuvo varios Nóbel relacionados con este campo. Edward C. Kendall, descubridor de la tiroxina. Encontró luego la “Cortina” cristalizada en 1934. Y un año después aisló la cortisona o Compuesto. Philip Hench, reumatólogo que al igual que Kendall trabajaba en la Clínica Mayo. Usó el denominado compuesto E en Artritis Reumatoide en 1948.
En la década de los 30 el europeo Thadeus Reichstein realizó numerosas investigaciones que llevaron al descubrimiento del compuesto Fa/E. De la adrenosterona y del núcleo pregnano. De la síntesis de la DOCA y de 40 esteroides. 6 de los cuales resultaron activos.
Otros investigadores encontraron las células yuxta-glomerulares. (Ruyter, 1935, Gourmaghtigh 1939). La aldosterona (Grundy, 1952) que inicialmente se llamó Electrocortina. La que causaba una reabsorción distal de sodio, excreción de potasio e hidrogeniones. Se vio también que la hipofisectomía no frenaba la producción de aldosterona. Pero que la nefrectomía no la dejaba actuar. En 1957, Braun-Menéndez y Schearz encontraron la angiotensina.
El siglo XX también aportó muchos conocimientos en la clínica. El neurocirujano Harvey Cushing describió el hipercortisolismo en 1932. Bartter, Bongiovani y Biglieri describieron varias deficiencias enzimáticas en los casos de hiperplasia suprarrenal congénita. Conn publicó sus casos de hiperaldosteronismo primario. Y Selye el síndrome general de adaptación o estrés, consistente en alarma, resistencia y agotamiento.
Referencias seleccionadas
- Aristizábal D. Fisiopatología de la hipertensión esencial. En: Cardiología (RH Rozo, A Merchàn y cols., Eds), Sociedad Colombiana de Cardiología 2000. pp.365-373.
- Biglieri EG, Arteaga E, Kater CE. Effect of ACTH on aldosterone and other mineralocorticoid hormones. In “Hypothalamic-pituitary-adrenal axis revisited. (WF Ganong, Ed.) Ann NY Acad Sci 1987. 512:426-437.
- Chiang JYL. Bile acid regulation of gene expression, role of nuclear hormone receptors. Endocr Rev 2002. 23: 443-463.
- Cody R. The integrated effects of angiotensin II. Am J Cardiol 1997. 79: 9-11.
- Dallman MF, Akana SF et al. Characterization of corticosterone feed-back regulation of ACTH secretion. In “The hypothalamic-pituitary-adrenal axis revisited” (WF Ganong, Ed.). Ann NY Acad Sci 1987. 512: 402-414.
- De Kloet ER, Ratka A et al. Corticosteroid receptor types in brain, regulation and putative function. In “The hypothalamic-pituitary adrenal axis revisited”(WF.Ganong,Ed.) Ann NY Acad Sci 1987. 512: 351-361.
- Goldfien A. Adrenocorticosteroids and adrenocortical antagonists. In “Basic and Clinical Pharmacology” (Katzung BG, Ed). Appleton-Lange, 1998. Pp.635-650.
- Katz A. Molecular biology of calcium channels in the cardiovascular system. Am J Cardiol 1997. 80: 171-221.
- Laragh JH, Sealy JE. The renin-aldosterone axis for blood pressure, electrolyte homeostasis, and diagnosis of high blood pressure. In: Textbook of endocrinology (RH Williams,Ed). WB Saunders, Philadelphia, 1981. Pp.1064-1079.
- Liddle G. The adrenals. In: Textbook of endocrinology (RH William, Ed.). WB Saunders, Philadelphia, 1981. Pp. 249-292.
- López-Jaramillo P. Bioquímica del endotelio vascular, implicaciones fisiológicas y clínicas. Horizonte Impresores, Bogotá, 5ª. Edición, 2001.Pp. 59-73.
- Miller WL. Molecular biology of steroid hormone synthesis. Endocr Rev 1988. 9: 295-318.
- Mulrow PJ,Takagi M et al. Inhibition of aldosterone secretion by atrial natriuretic peptide. In “Hypothalamic-pituitary –adrenal axis revisited” (WF Ganong, Ed.) Ann NY Acad Sci 1987. 512: 438-448.
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory and preparative actions. Endocr Rev 2000. 21: 55-89.
- Stein BC, Levin RI. Natriuretic peptides, physiology, therapeutic potential, and risk stratification in ischemic heart disease. Am Heart J 1998. 135. 914-923.
- Truss M, Beato M. Steroid hormone receptors, interaction with deoxyribonucleic acid and transcription factors. Endocr Rev 1993. 14:459-479.
- Varela E, Principios de biología molecular en el sistema cardiovascular. En: Cardiología (RH Rozo, A. Merchán y cols, Eds.). Soc Col Cardiol 2000. Pp.18-35.
- Williams GH, Dluhy RG. Diseases of the adrenal cortex. In “Harrison’s Principles of Internal Medicine”, 14th Ed. (Isselbacher KJ et al, Eds). McGraw-Hill 1998. Pp.2035-2056.