En el caso expuesto encontramos el desarrollo de infecciones recurrentes muy severas en el primer año de edad en una niña que había estado sana durante el primer semestre de vida.
La asociación etiológica de gérmenes intracelulares y extracelulares con niveles bajos en las cifras de anticuerpos séricos configura el cuadro correspondiente a una IDSC.
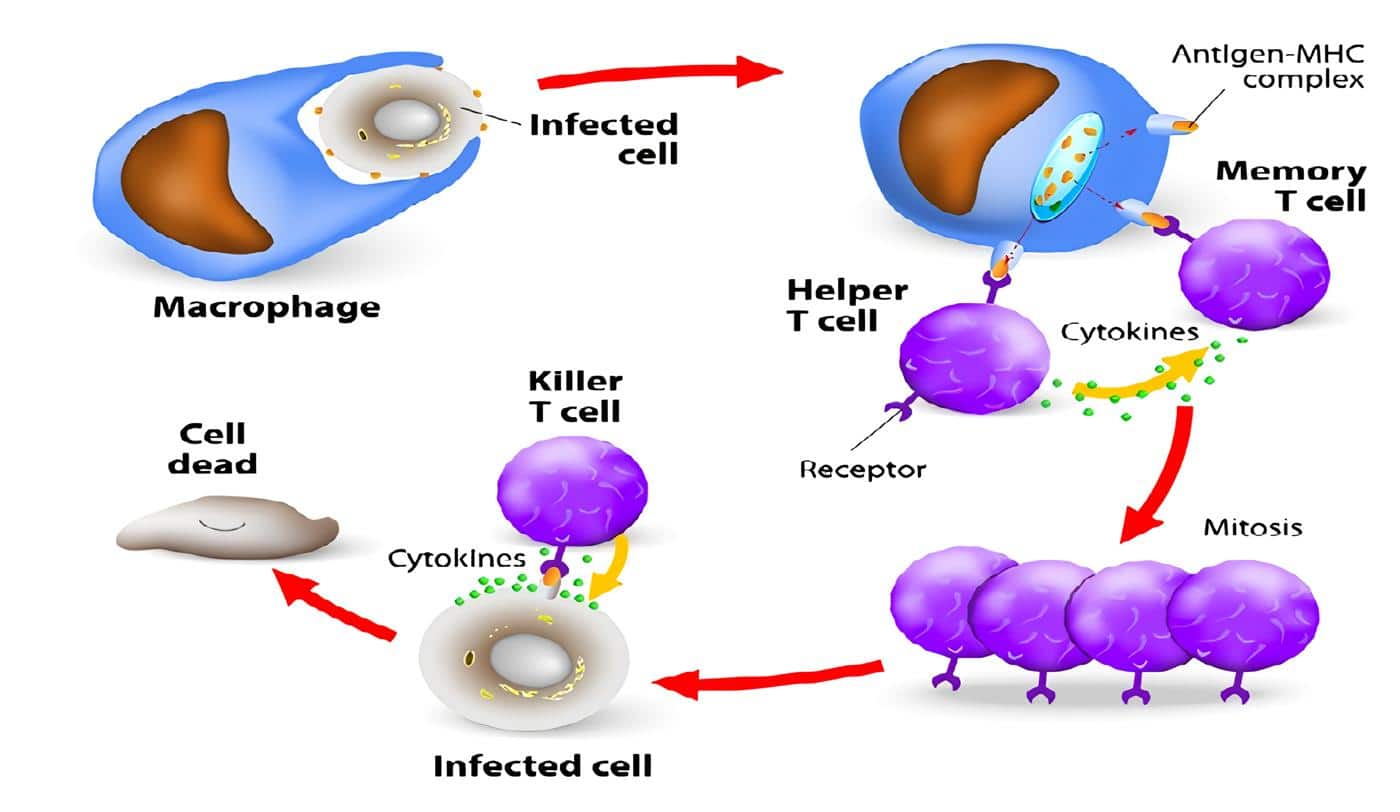
La IDSC es un síndrome de pronóstico reservado, gene-ralmente fatal en los dos primeros años de vida; aglutina un grupo de enfermedades caracterizadas por una marcada deficiencia en la función de los linfocitos T y B.
Está conformada por varias entidades de origen genético diverso, y en algunos casos la alteración molecular es aún desconocida.
A pesar de las diferentes causas hay rasgos muy constantes comunes a los diferentes tipos de IDSC, como son: elevada susceptibilidad al desarrollo de infecciones recurrentes severas, falla en el desarrollo del timo, linfopenia (desde leve hasta marcada), anormalidad en el desarrollo pondoestatural, candidiasis mu-cocutánea recurrente, diarrea persistente y neumonía generalmente intratable (principalmente por P. carinii). La IDSC afecta más a los varones (3:1) y se observa en los diferentes grupos étnicos.
Subclasificaciones al interior de la IDSC basadas en hallazgos fenotípicos
En la última década han existido notables avances en la caracterización de los defectos responsables de estas entidades; ésto ha originado subclasificaciones al interior de la IDSC basadas en hallazgos fenotípicos de los linfocitos circulantes y el patrón de herencia encontrado, permitiendo proponer dos grupos principales:
IDSC T(-)B(-)
IDSC T(-)B(-), Con disminución notable en los linfocitos T y B; afecta igual a ambos sexos pues los casos conocidos tienen un patrón de herencia autosómico recesivo. Las células NK pueden encontrarse en número normal o estar elevadas.
Las entidades caracterizadas en este grupo son la deficiencia de adenosín-deaminasa (ADA), deficiencia de purina nucleósido fosforilasa (PNP), la disgenesia reticular y la deficiencia de las enzimas recombinasas RAG1 y RAG2.
IDSC T(-)B(+):
Con bajo número de linfocitos T pero normal o elevado de linfocitos B; las células NK generalmente están ausentes.
Según el patrón de herencia se encuentran dos clases de IDSC T(-)B(+) que tienen una expresión clínica y un fenotipo exactamente igual, la deficiencia de la cadena gama común del receptor de IL-2 (forma ligada al cromosoma X, cerca del 50% de todos los tipos de IDSC) y la deficiencia de la tirosín kinasa Janus kinase 3 (JAK3) (autosómica recesiva).
La cadena gama común esta expresada en los receptores de las citoquinas IL-2, IL-4, IL-7, IL-9 e IL-15, y es la encargada de iniciar la transducción de las señales generadas por la unión con la citoquina correspondiente, las cuales son necesarias para la diferenciación, maduración y activación de diferentes tipos de linfocitos.
Después de la interacción en la superficie celular con alguna de las citoquinas mencionadas, la cadena gama común transmite una señal intracelular de activación por medio de su asociación con la tirosín kinasa JAK3, la cual una vez fosforilada se encarga de continuar con la transducción de los mensajes fosforilando factores de trans-cripción del grupo STAT, los cuales forman dímeros y migran al interior del núcleo celular para modificar el programa de transcripción de la célula.
La expresión del JAK3 está muy restringida a las células de la línea hematopoyética, aunque no exclusivamente a los linfocitos T (también está en células NK y linfocitos B); su papel es fundamental en la diferenciación de los distintos linfocitos, aunque se ha observado que la IL-4 puede activar la diferenciación de los linfocitos B por una vía independiente de Janus kinase 3 (JAK3).
Los primeros casos de IDSC debidos a mutaciones en Janus kinase 3 (JAK3):
Fueron informados en 1995 y desde entonces se ha ido constituyendo en una de las causas más importantes de LA IDSC autosómica recesiva.
La confirmación de la sospecha de inmunodeficiencia en esta paciente solo requirió evaluar los niveles séricos de inmunoglobulinas y el recuento de las diferentes subpoblaciones de linfocitos en sangre periférica; estos estudios de la-boratorio se encuentran disponibles en la mayoría de las ciudades grandes del país y están incluidos dentro de los estudios amparados por los diferentes planes de salud.
La caracterización molecular de éste y otros defectos inmunológicos se realiza hace algunos años por un grupo especializado en el diagnóstico y manejo de las inmunodeficiencias primarias localizado en el Laboratorio de Inmunología de la Universidad de Antioquia, el cual tiene la capacidad de brindar asesoría y apoyo en la evaluación de los pacientes con sospecha de inmunodeficiencias primarias.
Una vez realizado el diagnóstico, la paciente se ha incluido en un programa de manejo integral, con educación permanente sobre la necesidad de controlar la exposición a fuentes de infección, la nutrición y su medio ambiente; la terapia específica contempla quimioprofilaxis para prevenir las infecciones por P. carinii y gamaglobulina humana intravenosa mensual.
Todo lo anterior ha permitido que la paciente y su familia lleven una calidad de vida aceptable, debido a la disminución de los episodios infecciosos y la afortunada ausencia de nuevas hospitalizaciones.
El tratamiento ideal establecido para la IDSC es el transplante de médula ósea, con óptimos resultados cuando es HLA idéntico (sobrevida > 95%), y un poco menor pero infinitamente superior a la evolución natural de la enfermedad cuando es HLA haploidéntico (60%).
Transplante Haploidéntico
La paciente reportada aquí es hija única lo que, sumado a la carencia de un banco de donantes tipificados en nuestro medio, imposibilita la consecución de una médula HLA idéntica. Se podría pensar en un transplante haploidéntico tomando como donante a alguno de sus padres; sin embargo, la posibilidad de una reacción de injerto contra huésped, la necesidad de procesar la médula donante para eliminar los linfocitos T maduros, y la falta de grupos con experiencia en el transplante de médula ósea para individuos con IDSC hacen imposible por ahora alcanzar esta terapia en nuestro medio.
Finalmente, queremos resaltar como el enfoque integral e interdisciplinario de los pacientes con inmunodeficiencias primarias, aún en casos tan severos como la IDSC, permite cambiar la calidad de vida de pacientes y familiares y hacer un impacto notable en los costos y calidad de atención de los individuos afectados.
Referencias
- 1. Paul ME, Shearer WT. Approach to the evaluation of the immunodeficient patient. In: Rich RR, Fleisher TA, Schwartz BD, eds. Clinical Immunolgy: Principles and Practice. St. Louis: Mosby, 1996:609-620.
- 2. Salgado H, Montoya CJ, Henao J, Orrego JC, Patiño PJ. Infec-ción recurrente: evaluación de la respuesta inmune. Tópicos de Infectología 1999, pp. 161-169.
- 3. Comans-Bitter WM, de Groot R, Van den Beemad R, et al. Immunophenotyping of blood lymphocyte in childhood. Reference values for lymphocyte subpopulations. J Pediatr, 1997; 130: 388-393.
- 4. Candotti F, Villa A, Notarangelo LD. Severe Combined Immunodeficiency Due to Defects of JAK3 Tyrosyne Kinase. En: Primary immunodeficiency diseases. A molecular and gene-tic approach. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford, 1999. pp: 111-120.
Bibliografía
- 5. Buckley RH, Schiff IR, Schiff SE, Markert ML, Williams LW, Harville TO, Roberts JL, Puck JM. Human severe combined immunodeficiency: Genetic, phenotypic, and functional diversity in one hundred eigth infants. J Pediatr 1997;130:378-387.
- 6. Salgado H, Montoya CJ, Henao J, Orrego JC, Patiño PJ. Infección recurrente de origen inmunológico: atención del paciente. Tópicos de Infectología 1999, pp. 171-179.
- 7. Stiehm ER. Conventional therapy of primary immunodeficiency diseases. En: Primary immunodeficiency diseases. A molecular and genetic approach. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford, 1999. pp: 448-458.