Luego del diagnóstico específico el paciente recibió terapia permanente con Trimetoprín-Sulfa a dosis terapéutica cada 12 horas; por el aislamiento de Candida albicans de las lesiones cutáneas se agrega tratamiento con Ketoconazol por dos semanas, para continuar con una profilaxis tres días a la semana (lunes, miércoles y viernes).
A los 18 meses de vida presenta un cuadro progresivo de fiebre alta, compromiso del estado general, ictericia, distensión abdominal y hematoquexia; es hospitalizado y se realiza un diagnóstico de sepsis. En varios hemocultivos se aísla Salmonella spp.
Luego de 21 de tratamiento antibiótico (Cefotaxime y Prostafilina) es dado de alta, continuando su manejo permanente con Trimetropín-Sulfa.
A los 19 meses se diagnostica una bronconeumonía, se toman muestras para hemocultivos y se inicia manejo con Cefuroxima; el cuadro clínico progresa rápidamente, el paciente se deteriora y fallece luego de presentar un paro cardiorrespiratorio. Los resultados del hemocultivo reportaron Salmonella spp.
Comentario
Llama la atención en la historia de este paciente los antecedentes familiares, pues 3 hermanos de sexo masculino habían padecido de infecciones recurrentes severas y muerte en el primer años de vida.
Desafortunadamente, el desconocimiento en nuestro medio de las inmunodeficiencias primarias llevó a que el personal médico no sospechara una alteración de la inmunidad ligada al sexo como causa de su cuadro clínico y las defunciones.
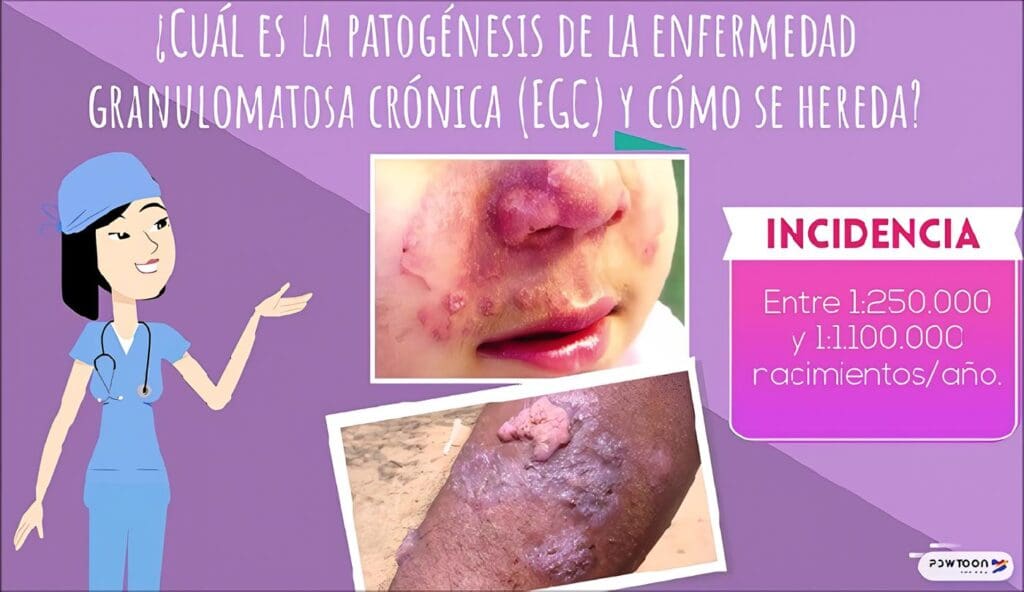
En este caso en particular es característica la aparición temprana de múltiples lesiones nodulares perianales recurrentes y resistentes al tratamiento que llevaron a fístulas perianales recidivantes; también se observó el desarrollo de infecciones micóticas en piel.
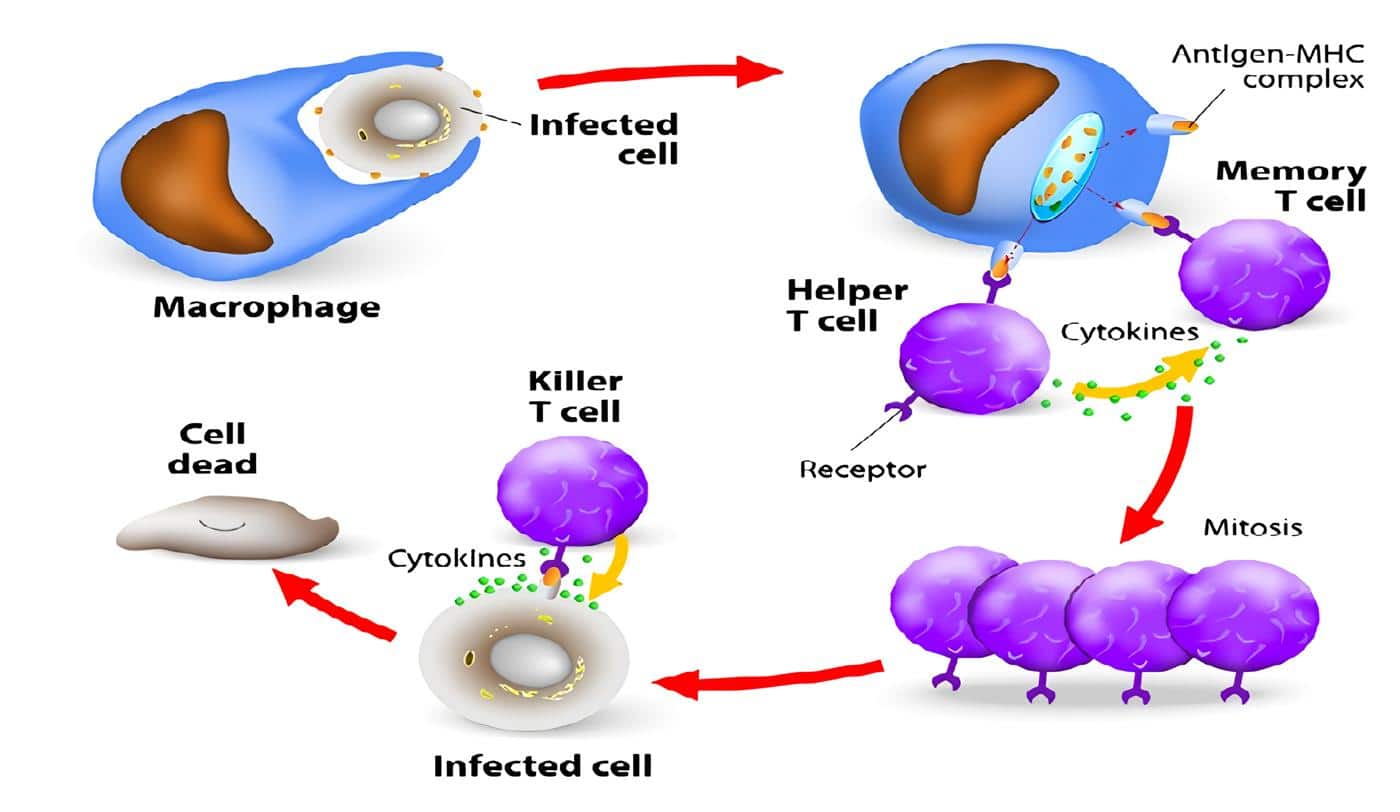
Lo anterior configuraba un cuadro altamente sugestivo de una deficiencia en la inmunidad mediada por las células fagocíticas.
La principal característica clínica de la Enfermedad Granulomatosa Crónica (EGC) es la aparición de las manifestaciones infecciosas antes del primer año de vida, constituyendo un síndrome de infección recurrente anormal que compromete la piel, el aparato gastrointestinal, el aparato respiratorio, los ganglios y otros órganos profundos como el hígado.
Son frecuentes la neumonía, la linfadenitis, los abscesos de piel y tejidos profundos, las fístulas perirrectales, y la septicemia.
Hallazgos no infecciosos como hepatoesplenomegalia y adenopatías múltiples también se observan con frecuencia.
Los microorganismos más frecuentemente implicados en las infecciones son los catalasa positivos, en especial el Staphylococcus aureus, Escherichia coli, Salmonella spp, Klebsiella spp, Bulkholderia cepacia y Aspergillus spp.
En este paciente no se realizó una búsqueda adecuada de los gérmenes durante la mayoría de las infecciones y por ello solo al final (en un centro de atención de tercer nivel) se pudo documentar la presencia de Salmonella.
La EGC defecto genético de baja incidencia
La EGC es un defecto genético de baja incidencia (1 en 250.000 individuos) traducido en una incapacidad de todas las células fagocíticas de la serie mielóide (neutrófilos, eosinófilos, monocitos y macrófagos) para generar los radicales reactivos del oxigeno producidos normalmente durante la explosión respiratoria de estas células.
El daño se encuentra a nivel de cualquiera de las proteínas que conforman la NADPH oxidasa, enzima encargada de la explosión respiratoria.
Esta enzima está compuesta por cuatro subunidades, dos localizadas en la membrana celular y de los gránulos secundarios (denominadas gp91 phox y p22 phox, en conjunto llamadas flavocitocromo b 558), y dos proteínas ubicadas en el citosol (p47phox y p67phox).
El defecto molecular en la EGC es bastante heterogéneo pues la enfermedad se origina a partir de mutaciones en cualquiera de los cuatro genes que codifican para las proteínas del sistema.
Así, mutaciones localizadas en el gen que codifica para la gp91phox (ubicado en el cromosoma X, Xp21-1) producen la variedad ligada al sexo, que afecta al 60% de todos los pacientes; el otro 40% de los casos se trasmite como rasgo autosómico recesivo pues los genes que codifican para p47phox, p67 phox y p22 phox se encuentran en los cromosomas somáticos.
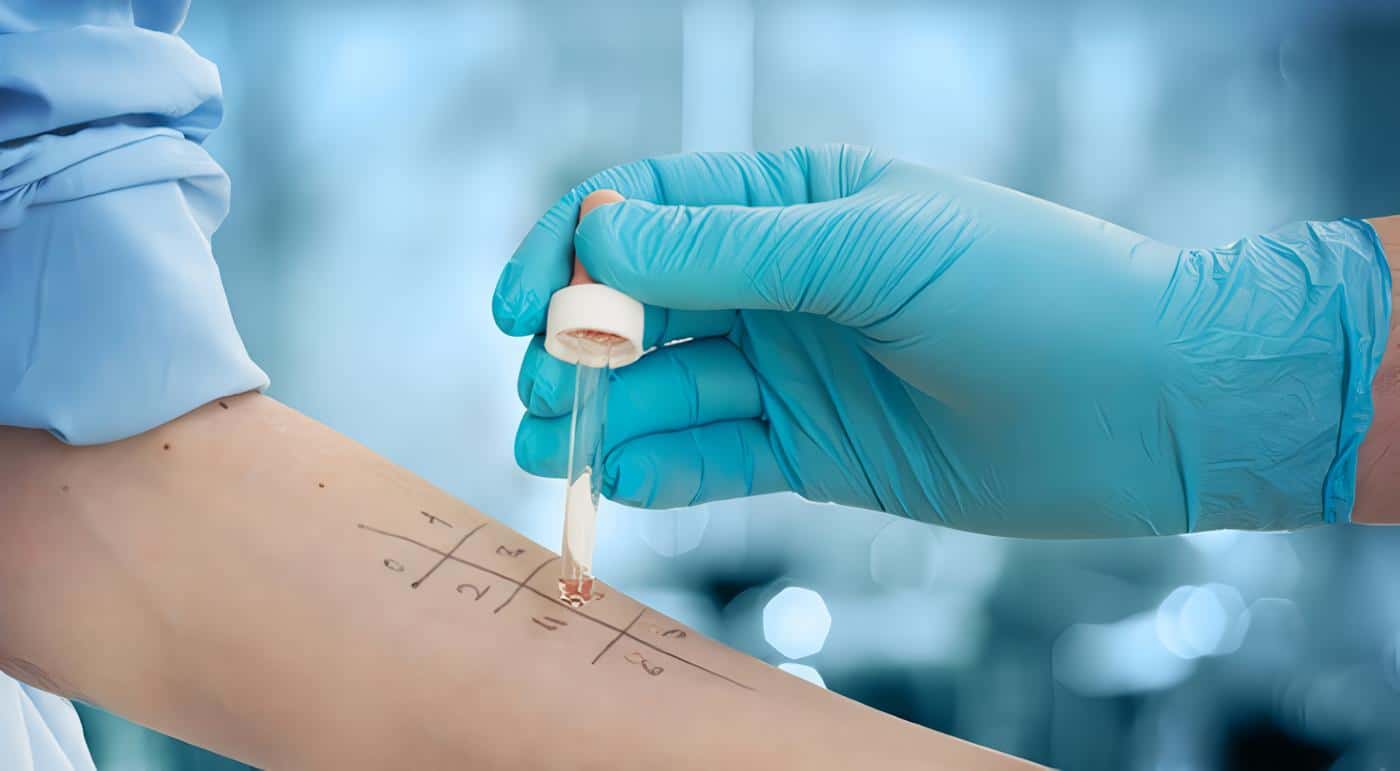
El diagnóstico de la EGC es bastante sencillo, pues existe una prueba bastante sensible y específica denominada “Reducción del NBT por neutrófilos en placa”, en la cual un cambio de color en el NBT indica que la activación de los neutrófilos estimulados con PMA ha conducido a la activación del sistema NADPH oxidasa y la generación de radicales reactivos derivados del oxígeno.
Una prueba confirmatoria más elaborada se hace por medio de citometría de flujo utilizando la Dihidrorodamina 123.
Caracterización molecular
La caracterización molecular de los defectos en cada paciente y su familia ya demanda de un centro especializado para su desarrollo. El Grupo de Inmunodeficiencias Primarias de la Universidad de Antioquia es un centro internacional de referencia para el estudio de la EGC y puede prestar los servicios de asesoría y apoyo para el diagnóstico y caracterización de los defectos a cualquier institución del país.
En el caso aquí presentado, el diagnóstico precoz de la enfermedad en los primeros niños afectados hubiese permitido detectar el estado portador de la madre para brindar consejería genética y ginecológica, destinada a escoger artificialmente el sexo de los hijos o hacer diagnóstico prenatal.
De igual forma, detectar la enfermedad en los primeros meses de vida permitiría instaurar un manejo interdisciplinario más adecuado que garantizara una mejor calidad de vida y mayor sobrevida; actualmente, varios centros internacionales tienen un protocolo de manejo con interferón gama recombinante que ofrece aceptables resultados en el control de la enfermedad.
En nuestro medio no se dispone de grupos que realicen transplante de médula ósea para pacientes con inmunodeficiencias primarias; en la medida que los pacientes se detecten en forma más temprana y sean mejor manejados será necesario implementar este tipo de acercamientos terapéuticos en Colombia.
Referencias
- 1. García de O D, Patiño PJ, Salgado H, López JA, Montoya CJ, Pérez JE. Evaluación del paciente con inmunodeficiencia. Síndrome de infección recurrente patológica. Medicina y Laboratorio 1997; 7: 545-575.
- 2. Holland MS, Gallin JI. Evaluation of the patient with suspected immunodeficiency. In: Mandell, Douglas, Bennett, eds. Principles and Practice of Infectious Diseases. New York: Churchill Livingstone, 1995: 149-158.
- 3. Curnutte JT. Chronic Granulomatous Disease: The solving of a clinical riddle at the molecular level. Clin Immunol Immunopathol 1993; 67: S2-S15.
- 4. Segal AW. The NADPH oxidase and chronic granulomatous disease. Molecular Medicine Today, 1996 (March): 129-135.
- 5. Roos D, Curnutte JT. Chronic Granulomatous Disease. En: Primary immunodeficiency diseases. A molecular and genetic approach. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford, 1999. pp: 353-374.
- 6. Salgado H, Montoya CJ, Henao J, Orrego JC, Patiño PJ. Infección recurrente de origen inmunológico: atención del paciente. Tópicos de Infectología 1999, pp. 171-179.
- 7. Stiehm ER. Conventional therapy of primary immunodeficiency diseases. En: Primary immunodeficiency diseases. A molecular and genetic approach. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford, 1999. pp: 448-458.