Citoquinas
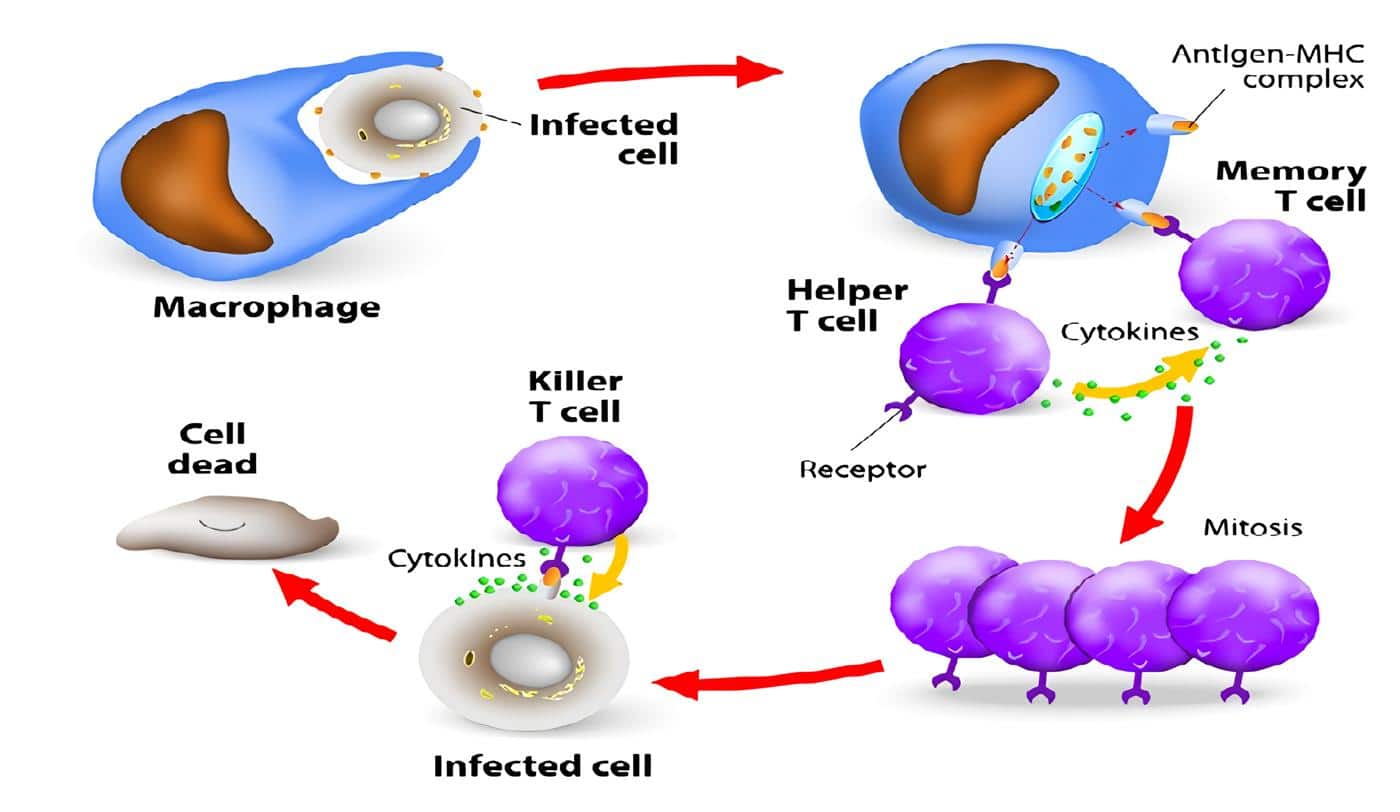
Las citoquinas son mediadores fundamentales de la comunicación intercelular aparecen como un nuevo grupo terapéutico en varias disciplinas. En oncología se utilizan IL-2, interferones (INFs), factor de necrosis tumoral (TNF) entre otros, para el manejo de leucemias y tumores sólidos avanzados.
Los defectos de la hematopoyesis se utilizan la eritropoyetina, el GM-CSF y el G-CSF.
En las enfermedades virales como las hepatitis B y C tienen utilidad los INFs, y en el SIDA se están llevando a cabo ensayos clínicos con IL-2.
En el campo de las inmunodeficiencias primarias existen varias patologías que en la actualidad son susceptibles de ser manejadas con varias citoquinas.
La inmunodeficiencia común variable se caracteriza por niveles subnormales de inmunoglobulinas, defecto en la producción de anticuerpos específicos e infecciones recurrentes por gérmenes, principalmente extracelulares. En estos pacientes no hay patrón de herencia definido.
Aunque la patógenesis de esta patología no se ha definido, en la mayoría de los casos la ICV se presenta como una alteración multisistémica de la inmunorregulación, en la cual aparecen con mucha frecuencia manifestaciones de autoinmunidad.
Desde hace varios años se viene utilizando la interleuquina-2 (IL-2) para el manejo de esta enfermedad con resultados bastante prometedores.
En un paciente con inmunodeficiencias severa combinada que presentaba un defecto en la síntesis de IL-2 reveló una mejoría de la inmunidad después de la administración de esta citoquina.
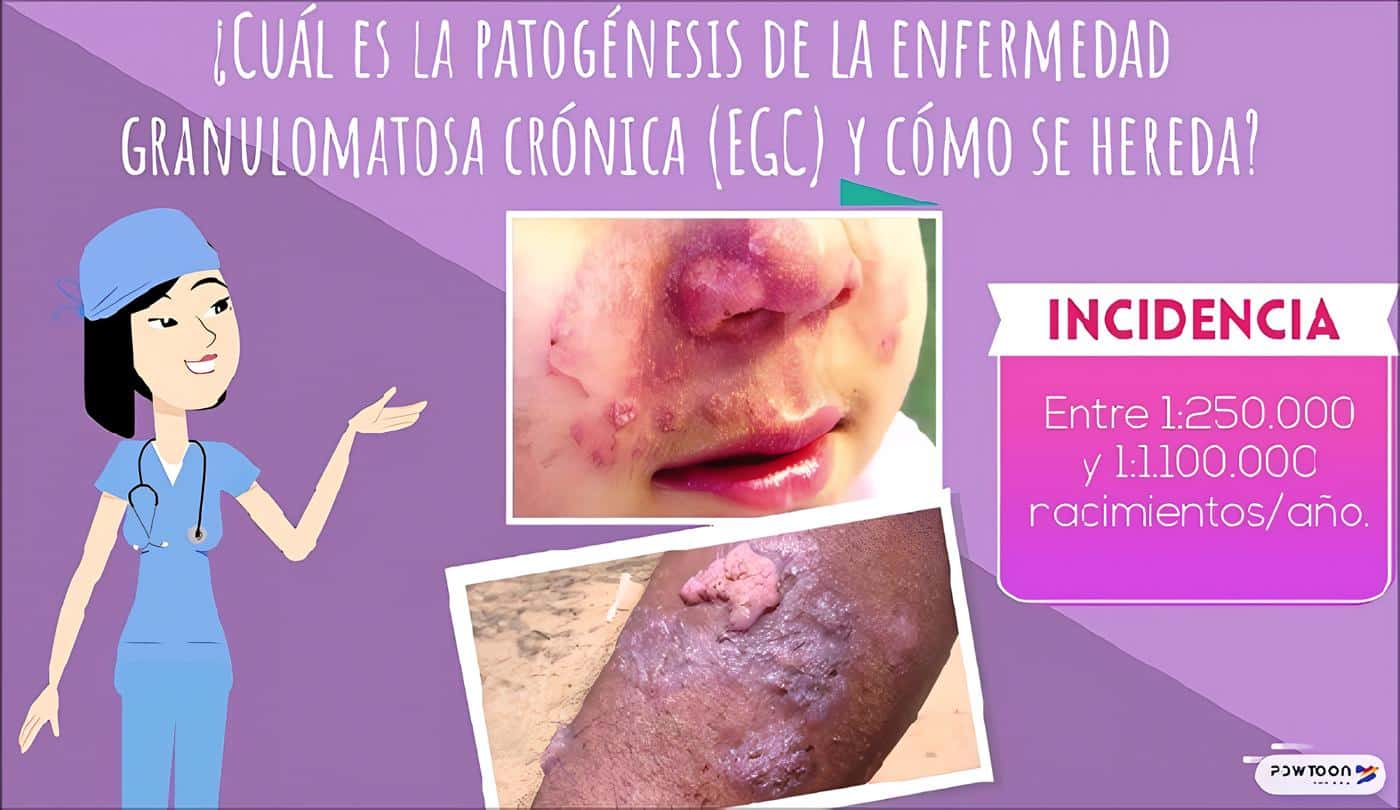
Por su parte, la única aplicación terapéutica que se ha aprobado por la FDA para el INF-g es en el manejo de la enfermedad granulomatosa crónica (EGC).
La EGC se debe a un defecto en la explosión respiratoria de las células fagocíticas:
En el cual los pacientes no producen los metabolitos de oxígeno necesarios para la destrucción de una gran variedad de gérmenes. El defecto genético esta localizado en uno de cuatro genes que codifican para las proteínas que conforman el sistema transportador de electrones conocido como NADPH oxidasa.
El efecto benéfico del INF-g ha sido claramente establecido, especialmente cuando se administra en forma simultánea con antibióticos profilácticos; sin embargo el mecanismo por el cual esta citoquina actúa en estos pacientes no está claramente establecido. El INF-g aumenta la expresión de los genes de la NADPH oxidasa.
También se ha propuesto que esta citoquina la producción de óxido nítrico por las células fagocíticas, el cual tiene potente actividad microbicida; aunque ésto es cierto en el ratón, en el humano no hay evidencias claras de que este fenómeno este ocurriendo.
Desde hace algún tiempo se ha demostrado que el INF-g aumenta la expresión del receptor Fc de las células fagocíticas, el cual es esencial para la fagocitosis de microorganismos opsonizados. Ahora se acepta que el aumento de la capacidad fagocítica en combinación con el potenciamiento de sistemas microbicidas independientes del oxígeno sean los mecanismos de acción principales del INF-g en el manejo de la EGC.
Además de la EGC existen otros defectos inmunológicos que se pueden beneficiar del uso del INF-g. Hace poco se describió una familia en la cual varios miembros presentaban una infección diseminada por el Mycobacterium avium.
El análisis inmunológico demostró que:
Estos pacientes presentaban un defecto en la producción de IL-12 por parte de los monocitos durante la presentación antigénica al linfocito T, lo cual se caracteriza por la incapacidad de estos linfocitos para producir INF-g en respuesta al estímulo antigénico.
Lo anterior, además de demostrar el papel importante de la IL-12 en la respuesta inmune a gérmenes intracelulares, abre la posibilidad del tratamiento con IL-12 sola o en combinación del INF-g en aquellos defectos de la respuesta inmune a este tipo de microorganismos.
La neutropenia crónica severa es un grupo heterogéneo de enfermedades caracterizadas por una disminución selectiva en el número de neutrófilos circulantes a niveles que con frecuencia se asocian fiebre recurrente, inflamación orofaríngea crónica e infecciones severas.
La neutropenia crónica severa se divide en tres formas: neutropenia idiopática, neutropenia cíclica y formas congénitas de neutropenia.
Para el manejo de estas patologías se cuenta ahora con dos citoquinas: los factores estimuladores de colonias de granulocitos-monocitos (GM-CSF) y de granulocitos (G-CSF).
El G-CSF (filgrastim) es un factor de crecimiento hematopoyético que tiene la capacidad de promover la proliferación y maduración de células mieloides y en particular la proliferación y diferenciación de neutrófilos, tanto in vivo como in vitro. Los resultados de los ensayos de fase III del uso del G-CSF en pacientes con diferentes formas de neutropenia crónica revelan su utilidad para corregir los niveles de neutrófilos circulantes.
Trasplante de médula ósea
En la actualidad el único tratamiento causal de las inmunodeficiencias primarias es el trasplante de médula ósea.
Este procedimiento puede revertir la anormalidad inmunológica y curar definitivamente a los pacientes.
Un factor importante para el pronóstico favorable es la condición clínica del paciente al momento del trasplante, por lo tanto mientras más temprano se establezca el diagnóstico de algún defecto inmunológico mucho mejor será el pronóstico si se decide realizar un trasplante de médula ósea.
A pesar de los avances recientes en el trasplante de médula ósea el porcentaje de éxito en las inmunodeficiencias primarias sigue siendo limitado.
Son varios los factores que frustran las expectativas de éxito del trasplante de médula ósea en estos pacientes, pero principalmente la enfermedad injerto versus huésped (EIvsH) que se produce cuando las células inmunocompetentes de la médula ósea trasplantada atacan a las células del receptor y las infecciones por gérmenes oportunistas que aparecen como consecuencia de la ablación de médula ósea y de la inmunosupresión, sea para evitar el rechazo o la EIvsH.
La correción inmunológica con el trasplante de médula ósea se ha practicado en muchas de las inmunodeficiencias primarias, pero especialmente en la inmunodeficiencia severa combinada y el síndrome de Wiskott-Aldrich.
Recientemente, Rebeca Buckley ha recopilado la información pertinente a los trasplantes de médula ósea en pacientes con inmunodeficiencia primaria realizados en diferentes centros del mundo en un período de 26 años y reporta una sobrevida del 62%.
Aunque esta cifra parece ser alta, es necesario entrar a considerar cuales de los pacientes en realidad tienen establecimeinto del injerto y por lo tanto corrección del defecto inmunológico.
Terapia génica
El reemplazo de un gen defectuoso es el sueño de todos aquellos que tenemos que ver con pacientes que presentan un defecto hereditario.
En teoría, cualquier defecto genético que pueda ser corregido con un trasplante de médula ósea alogénico es un candidato potencial para una terapia génica dirigida a corregir las células madre multipotenciales hematopoyéticas.
La dificultad para encontrar donantes histocompatibles y la alta frecuencia de EIvsH después del trasplante de médula ósea en las inmunodeficiencias primarias han hecho necesario el desarrollo de la terapia génica como alternativa para lograr una corrección definitiva de estas patologías.
Un requisito fundamental para que un defecto sea susceptible de corrección genética es el de que el gen comprometido haya sido identificado y clonado.
Se han realizado varios ensayos clínicos de terapia génica en algunas inmunodeficiencias y en otras ya existen modelos in vitro o en animales que hacen suponer que en poco tiempo se estén aplicando en el humano.
La deficiencia de adenosina deaminasa (ADA) fue la primera inmunodeficiencia en la que el defecto molecular fue caracterizado y la primera enfermedad hereditaria en la que se llevó a cabo un ensayo clínico de terapia génica.
Las células T de dos pacientes con esta enfermedad se ailslaron y se cultivaron in vitro con estímulos que indujeron una gran proliferación, luego estas células se expusieron vector retroviral que contenía el DNA correspondiente al gen de la ADA.
Después de 9 a 12 días de cultivo las células se administraron i.v. Este tratamiento se repitió en 11 o 12 ocasiones en un período de menos de 2 años. Dos años más tarde del fin de esta terapia ambas pacientes presentaban una reconstitución de las respuestas inmunes celular y humoral.
Más recientemente, en 3 recién nacidos, en los que el diagnóstico había sido hecho por amniocéntesis:
se hizó una corrección genénetica de células madre CD34+ obtenidas del cordón umbilical al momento del nacimiento.
La ISC ligada al cromosoma X se debe a mutaciones de la cadena gama que es común a los receptores para IL-2, IL-4, IL-7, IL-9 e IL-15.
Varios grupos de investigación han iniciado ensayos preclínicos de transferencia del gen de esta cadena gama a células aisladas de pacientes con ISC, los cuales demuestran una corrección de la respuesta proliferativa de los linfocitos T inducida por la IL-2.
Estos resultados hacen suponer que en poco tiempo será posible corregir este defecto en los pacientes con ISC ligada al sexo.
La EGC es otra inmunodeficiencia en la que la terapia génica es una realidad. Recientemente se describió el primer ensayo clínico en 3 pacientes con deficiencia de la p47-phox.
A células madre hematopoyéticas CD34+ se les hizo una transferencia del gen de dicha proteína utilizando un retrovirus, luego las células fueron reinyectadas a los pacientes en una sola dosis.
Las células fagocíticas de estos pacientes presentaron una reconstitución inicial de la capacidad de producir radicales de oxígeno, sin embargo después de un seguimiento de 6 meses, esa función desapareció.
Ésto demuestra la necesidad de la administración periódica de las células madre corregidas.
En las demás formas de EGC, especialmente en la deficiencia de gp91-phox, los ensayos preclínicos están en etapas muy avanzadas y es de esperar su aplicación en el humano en poco tiempo.
Conclusión
Como se puede observar el manejo de las inmunodeficiencias primarias puede ir desde medidas relativamente sencillas, especialmente dirigidas a que los pacientes conozcan su problema y lo puedan manejar en forma autosuficiente, hasta formas de tratamiento altamente especializado y aún en etapas experimentales que está disponible solo en ciertos centros.
Es fundamental hacer un diagnóstico precoz de una inmunodeficiencia primarias, pues mientras más temprano se logre incidir sobre las complicaciones infecciosas de estos pacientes, en la mayoría de las veces con las medidas generales, mucho mejor será el pronóstico y la calidad de vida tanto de ellos como de sus familias.
Referencias
- 1. Blaese RM. Gene therapy: applications for immunodeficiency and cancer. En: Rich, RR, Fleisher TA, Schwartz BD, Shearer WT, Strober W. Eds. Clinical Immunology. Principles and Practice. St. Louis, Mosby, 1996, pp. 1931-1929.
- 2. Buckley RH. Bone marrow reconstitution in primary immuno-deficiency. En: Rich, RR, Fleisher, TA, Schwartz, B, Shearer, WT, Strober, W. Eds. Clinical Immunology. Principles and Practice. St. Louis, Mosby, 1996, pp. 1813-1830.
- 3. Cunningham-Rundles C, Kazbay K, Zhou Z, Mayer L. Immuno-logic effects of low-dose polyethylene glycol-conjugated recombinant interleukin-2 in common variable immunodeficiency. J Interferon Cytokine Res 153: 269-276, 1995.
- 4. Polmar SH, Sorensen R. Immunoglobulin replacement therapy in primary immunodeficiency diseases. En: Rich RR Fleisher TA, Schwartz BD, Shearer WT, Strober W. Eds. Clinical Immunology. Principles and Practice. St. Louis, Mosby, 1996, pp. 1865-1875.
- 5. Smith CIE, Ochs HD, Puck JM. Genetically determined immunodeficiency diseases: A perspective. In: Smith CIE, Ochs HD, Puck JM. Eds. Primary Immunodeficiency Diseases. A molecular and genetic approach. New York, Oxford University Press, pp 3-11, 1999.
- 6.Stiehm ER. Conventional therapy of primary immunodeficiency diseases. In: Smith CIE, Ochs HD, Puck JM. Eds. Primary Immunodeficiency Diseases. A molecular and genetic approach. New York, Oxford University Press, pp 448-458, 1999.