Felipe Solano1, Jorge de los Ríos2, Juan David Matute3, Wildeman Zapata4,
María Margarita Olivares, MD5, Pablo Javier Patiño, MD, DSc6, Carlos Julio Montoya, MD, MSc7.
1-3Estudiantes de medicina, Universidad de Antioquia
4Bacteriólogo, ASISTENTE de laboratorio Grupo de Inmunodeficiencias Primarias
5-7Grupo de Inmunodeficiencias Primarias (IDP), Corporación Biogénesis, Facultad de Medicina, Universidad de Antioquia,
Medellín – Colombia.
Correspondencia:
María Margarita Olivares G, MD.
Carrera 51 D, No. 62-29. Laboratorio de Inmunología
Facultad de Medicina – Universidad de Antioquia
Medellín, Colombia
Teléfonos: 510 60 57 – 510 60 78 Fax: 510 6047
e.mail: marguy@epm.net.co
La agammaglobulinemia ligada al cromosoma X (XLA) es una inmunodeficiencia primaria caracterizada por disminución de linfocitos B circulantes y por la reducción de los niveles plasmáticos de varios isotipos de inmunoglobulinas.
Los individuos afectados, tienen mayor susceptibilidad de presentar infecciones bacterianas del tracto respiratorio, así como infecciones por enterovirus.
En este reporte se describe la historia clínica de tres pacientes de sexo masculino vinculados al servicio clínico del grupo de inmunodeficiencias primarias de la Universidad de Antioquia, en quienes se estableció el diagnóstico de XLA. Gracias a esto los pacientes se beneficiaron de una terapéutica con gamaglobulina intravenosa, lo que ha mejorado sustancialmente su calidad de vida.
Palabras clave: agamaglobulinemia congénita ligada al X (XLA), inmunodeficiencia primaria, inmunoglobulinas.
Abstract
The X linked agammaglobulinemia (XLA) is a primary immunodeficiency characterized by absence of circulatory B lymphocytes and drastic reduction of plasmatic levels of several immunoglobuline isotypes.
The affected individuals have larger susceptibility to present bacterial infections of respiratory tract as well as infections by enterovirus.
This report describes the clinic history of three male patients enrolled to the Clinic Service of Primary Immunodeficiency Group at University of Antioquia in whom the diagnosis of XLA have been stablished. The therapy with intravenous gammaglobuline has improved substantially their quality of life.
Key words: The X linked agammaglobulinemia, primary immunodeficiency, immunoglobulines
Introducción
La agamaglobulinemia ligada al cromosoma X (XLA), descrita por Bruton en 1952, fue la primera entidad conocida como una inmunodeficiencia primaria. La XLA es una inmunodeficiencia humoral con una incidencia de cinco individuos afectados por cada millón de personas nacidas.
Es causada por mutaciones en el gen que codifica para la proteína BTK (Tirosina Kinasa de Bruton). Hasta el momento se han registrado 605 mutaciones, de las cuales un tercio son mutaciones de novo y el resto se presentan como casos familiares.
Se han reportado diferentes tipos de mutaciones las cuales incluyen cambios con sentido, alteraciones de sentido, deleciones e inserciones (1, 2).
La XLA se caracteriza por una profunda deficiencia de linfocitos B (LB), así como por disminución de los niveles plasmáticos de varios isotipos de inmunoglobulinas (principalmente IgG, IgM, IgA).
La proteína BTK es necesaria para la diferenciación y supervivencia de los LB, pues su ausencia conduce a un bloqueo en la maduración de células pro-B a células pre-B en la médula ósea. Igualmente, en estos pacientes se han descrito defectos en la maduración tardía de los LB (1, 3).
Puesto que la IgG es transportada activamente a través de la placenta:
Durante el último trimestre del embarazo, los niños con esta anomalía tienen niveles normales de IgG al nacimiento, sin embargo, disminuyen progresivamente.
Antes de su primer año de vida, estos pacientes tienen mayor susceptibilidad a infecciones bacterianas extracelulares; la resistencia a infecciones virales está intacta, a excepción por una inusual susceptibilidad a infecciones por enterovirus, lo cual pudiera resultar en polio paralítica o en meningoencefalitis asociada con la vacuna anti-polio de virus atenuados.
En los pacientes con esta enfermedad es frecuente encontrar hipoplasia de nódulos linfoides, lo cual se puede demostrar fácilmente observando el tamaño de las amígdalas, las cuales normalmente presentan un tamaño considerable en los períodos de lactancia tardía y PREESCOLAR.
Dentro de la enfermedad se han evidenciado varios fenotipos clínicos (Tabla 1), pero hasta el momento no se ha evidenciado una relación causal entre los fenotipos y los genotipos, incluso un mismo genotipo puede causar la forma clásica y la forma leve, situación que subraya en esta inmunodeficiencia la importancia de otros factores, tales como la influencia de otros factores genéticos y las situaciones ambientales en las que vive el paciente (1, 3).
Tabla 1. Fenotipos clínicos de la agamaglobulinemia ligada al X (XLA).
| Fenotipos de XLA | XLA severa (clásica) | XLA leve |
| Porcentaje de casos | ~97% | ~3% |
| Comienzo de síntomas | ~6 meses-1 año de edad | Algunos asintomáticos hasta adolescente |
| Edad promedio de diagnóstico | 2.5 años con historia familiar y 3.5 años sin historia familiar | Algunos en la adolescencia |
| Evolución de las infecciones | Infecciones recurrentes anormales en tracto respiratorio, gastrointestinal y piel; también pueden presentar meningitis y septicemia.Estas infecciones son causadas principalmente por bacterias extracelulares, enterovirus, Giardia lamblia, P. Carinii entre otros. | Infecciones con menor recurrencia y severidad que en la FORMAclásica. |
| Niveles de inmunoglobulina | La mayoría los tiene marcadamente disminuidos o ausentes | Parcialmente disminuidas o normales |
| Linfocitos B en sangre periférica | Menor del 2% | Algunos presentan valores hasta del 6 o 10% |
El gen de la BTK se encuentra en el cromosoma X:
En la región q21.33-q22 (Figura 1); tiene un tamaño de 37.5 kb y posee 19 exones, de los cuales 18 codifican para la proteína.
La transcripción genética se da en dirección telómero a centrómero y el producto de esta trascripción es una proteína citoplasmática tipo tirosina kinasa que es expresada en todas las líneas de células hematopoyéticas, excepto en linfocitos T y células plasmáticas.
Este grupo de proteínas, en especial las de la subfamilia “SRC”, a la que pertenece la BTK, se encuentran libres en el citosol o unidas a receptores de la membrana citoplasmática cumpliendo un rol de “transductores de señales” hacia el interior de la célula. Esta proteína se encarga de catalizar la fosforilación de residuos de tirosina en diferentes proteínas, empleando moléculas como el ATP y otros nucleótidos donadores de fosfatos. La proteína posee 659 aminoácidos y tiene un peso molecular de 77 kDa.
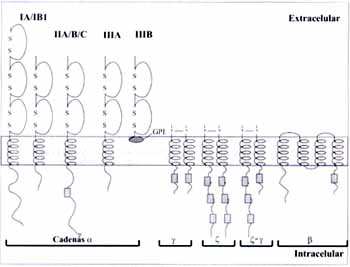
Desde el punto de vista funcional está compuesta por 5 dominios distintos: el dominio PH (“pleckstrin homology”) en la región N-terminal, seguido por el dominio TH (“TEC homology”) rico en prolina; el dominio SH3 (“Src homology 3”) que media la unión a proteínas con motivos ricos en prolina; el dominio SH2 (“Src homology 2”) que permite la interacción con motivos de tirosina fosforilada, y una región C-terminal catalítica o dominio SH1 que posee función kinasa (Figura 2) (1, 4).
")
Figura 1. Localización del gen que codifica para la tirosina kinsa de Bruton (BTK), el cual se encuentra ubicado en el cromosoma X. Extraída de: https://www.ncbi.nlm.nih.gov/htbin-post/Omim/getmap?l300300 (9).

Figura 2. Modelo esquemático que muestra los diferentes dominios de la BTK. PH: dominio de homología a plecstrina, SH: dominio de homología a SRC, TH: dominio de homología a TEC.
El diagnóstico de XLA se inicia por la realización de pruebas que confirman o identifican los defectos inmunológicos, las cuales incluyen medidas de inmunoglobulinas y el recuento de LB, sub-poblaciones de células T y células asesinas naturales (NK).
Para el análisis de mutaciones se pueden usar técnicas como la reacción en cadena de la polimerasa usando transcriptasa reversa (PCR-RT), polimorfismos conformacionales de cadena sencilla (SSCP), electrofóresis en gel con gradiente de desnaturalización (DGGE), corte químico de nucleótidos no apareados (CCM), inmunodetección con anticuerpos específicos, hibridación de Southern, entre otras técnicas (4).
Se puede diagnosticar la XLA prenatalmente a partir de líquido amniótico midiendo niveles de células B fetales o usando técnicas moleculares que identifican el defecto genético (1).
El diagnóstico diferencial de esta entidad se debe establecer con otras inmunodeficiencias primarias como son el déficit selectivo de IgA, en donde se encuentra una disminución de la IgA 1 y 2 pero los LB son normales; el síndrome de Hiper-IgM, en donde se encuentra un aumento de esta inmunoglobulina, con IgD aumentada o normal y disminución de los otros isotipos; las deficiencias selectivas de subclases de IgG, en las cuales hay disminución en una o más subclases de IgG.
La deleción de las cadenas pesadas de las inmunoglobulinas, que se presenta con ausencia de IgG 1, 2 ó 4, y en algunas ocasiones se asocia a la ausencia de IgA o IgE, además tiene un patrón de herencia autosómico; la hipogammaglobulinemia transitoria de la infancia, en esta se encuentran niveles disminuidos de IgG e IgA, niveles detectables de anticuerpos antibacterianos y LB normales; y por último con la inmunodeficiencia común variable, que se caracteriza por reducción variable en múltiples isotipos de inmunoglobulinas, con LB normales o ligeramente disminuidos (5).
El manejo de la XLA NECESITA de medidas generales:
Las cuales incluyen cambios radicales en el estilo de vida tanto personales como sociales, educación sobre la no exposición innecesaria a agentes infecciosos, una dieta balanceada que cubra las necesidades nutricionales, enseñar sobre lo perjudicial de consumir sustancias psicoactivas (ni activa ni pasivamente), explicar que estos pacientes no deben recibir vacunas vivas (como la de polio, triple viral; varícela-zoster o tifoidea oral).
Aunque las vacunas no replicativas son poco eficaces, puesto que estos niños no desarrollan memoria de LB, no existe mayor riesgo al aplicarles vacunas que contienen virus muertos o proteínas recombinantes; además éstas pueden ser potencialmente benéficas por la estimulación inmune, al inducir la memoria de LT (6).
En el manejo de un niño con XLA se debe administrar empíricamente, ante cualquier sospecha de infección activa, un antibiótico de FORMA temprana, y esta terapia debe ser completa y enérgica luego de establecer el germen responsable de la infección.
Está ampliamente descrito que los pacientes con XLA se benefician del tratamiento profiláctico mensual de forma indefinida con gamaglobulina humana intravenosa en una dosis de 400 mg por Kg cada 25 a 30 días (7).
Una vez se inicia la terapia con gamaglobulina se deben realizar seguimientos semestrales, midiendo IgG sérica y realizando pruebas de función pulmonar, hepática y renal, química sanguínea y Rx de tórax, para determinar si existe una reacción adversa a la terapia; estas revisiones se realizan con el fin de individualizar la terapia con gamaglobulina humana intravenosa buscando la dosis óptima y la frecuencia de la infusión para cada paciente (8).
El pronóstico de la enfermedad es variable:
Es así como los pacientes que no reciben tratamiento su tiempo de supervivencia promedio se ha calculado entre 3-8 años, mientras que los pacientes que son tratados adecuadamente el tiempo de supervivencia se prolonga hasta la tercera a cuarta década de la vida (1, 3).
En este artículo se expone la evolución clínica de tres pacientes que desde temprana edad presentaron un síndrome severo de infección recurrente, con compromiso de piel, sistema respiratorio superior e inferior y sistema gastrointestinal.
A pesar de este cuadro fueron remitidos en FORMA tardía para evaluación de su inmunocompetencia, después de lo cual se estableció el diagnóstico de XLA.
El desconocimiento de entidades, como son las inmunodeficiencias, y de su impacto sobre la salud individual y colectiva, pueden explicar la tardanza en la sospecha clínica, así como la negativa para suministrar la terapia de reemplazo indicada en esta situación particular.
(Lea También: Grupo de Inmunodeficiencias Primarias (IDP))
Presentación de los Casos
Caso número 1
Paciente de sexo masculino, remitido a los tres años y siete meses de vida para el ESTUDIO de un síndrome de infección recurrente anormal en el Grupo de Inmunodeficiencias Primarias (IDP) de la Universidad de Antioquia.
Sus manifestaciones clínicas se iniciaron al año y cinco meses de vida aproximadamente, con otitis media aguda supurada (OMA) y enfermedad diarreica aguda (EDA) a repetición, por lo cual fue hospitalizado y recibió tratamiento con amoxacilina y metronidazol. A los 20 meses de edad presentó nuevamente OMA, bronconeumonía (BNM) y conjuntivitis, por lo cual requirió ser hospitalizado y tratado con múltiples antibióticos.
A los 25 meses de edad se hospitalizó de nuevo y fue tratado con antibióticos por celulitis del pie izquierdo.
Luego de esto el niño es hospitalizado en varias ocasiones (dos veces por mes), con una duración que variaba entre tres y 15 días, por infecciones respiratorias, gastrointestinales y cutáneas.
En febrero de 2001 se estableció el diagnóstico de inmunodeficiencia primaria humoral y se inició el tratamiento con gamaglobulina humana intravenosa.
Este niño es producto del tercer embarazo, con una duración de la gestación de 9 meses y de buena evolución, el parto fue normal, con una talla y peso al nacer adecuados para la edad gestacional (54 cm y 3500 g) y sin anoxia perinatal.
La caída del cordón ocurrió en la primera semana de vida y recibió LACTANCIA durante aproximadamente un año y medio. El esquema de vacunación del PAI está completo para su edad.
Estrabismo corregido quirúrgicamente
El paciente tiene como otros antecedentes personales de importancia estrabismo corregido quirúrgicamente a los dos años y medio de edad y diagnóstico de luxación congénita de cadera, la cual fue tratada con arnés y silla especial hasta los 18 meses de edad.
Cuando se solicitó evaluación por neuropediatria por presentar episodios convulsivos, se realizó TAC y se diagnóstico esquisencefalia de labio abierto con hemiparesia izquierda, lo que descartó el diagnóstico luxación congénita de cadera. Para esto fue tratado con terapia física y de lenguaje; actualmente no recibe tratamiento neurológico con medicamentos.
Dentro de los antecedentes familiares se destaca un hermano que falleció en sus primeros años de vida con un cuadro de BNM, OMA y conjuntivitis a repetición con diagnóstico de inmunodeficiencia humoral; igualmente tiene un primo materno con diagnóstico de inmunodeficiencia de IgG, que actualmente es tratado con gamaglobulina humana venosa.
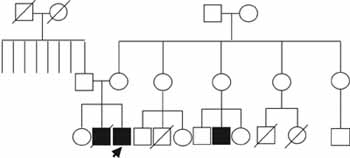
Además, los familiares refieren dos muertes de primos maternos a edades tempranas por infecciones. A parte de estos antecedentes se encontraron dermatitis alérgica en una hermana y asma en una prima paterna (Figura 3).

Figura 3. Genealogía del caso número uno. El patrón de herencia sugiere un rasgo ligado al cromosoma X. El caso índice se señala con una flecha.
Los exámenes realizados, desde la sospecha clínica de inmunodeficiencia primaria humoral en febrero del 2001 y antes de comenzar la terapia con gamaglobulina revelaron niveles de inmunoglobulinas séricas casi ausentes (IgE: 1 UI/ml, IgG 0.58 mg/dl, IgM < 0.034 mg/dl, IgA < 40.9 mg/dl). Luego de instaurar la terapia mensual con la dosis adecuada de gamaglobulina, se incrementaron los niveles paulatinamente, hasta que se logró mantener la concentración de IgG sérica estable (niveles de IgG que oscilaron entre 580 y 849 mg/dl).
Al llegar al servicio clínico del grupo de Inmunodeficiencias Primarias, se diagnosticó un síndrome de infección recurrente anormal, como consecuencia de una inmunodeficiencia humoral.
Con el fin de documentar las anormalidades inmunológicas, luego de realizar un examen físico detallado, que reveló una hemiparesia izquierda y amígdalas no visibles, se realizaron exámenes de laboratorio complementarios que incluyeron poblaciones de LT, LB y células NK, que evidenciaron unos LB casi ausentes en sangre periférica (Tabla 2).
Tabla 2.Valores de poblaciones de LT, LB y células NK del caso número uno.
| Descripción | Valor (%) | Células x ml | Valor esperado |
| Leucocitos | 7480 | ||
| Linfocitos | 30 | 2244 | 1700-6900 |
| LT CD3+ | 70 | 1571 | 900-4500 |
| LT CD4+ | 27 | 608 | 500-2400 |
| LT CD8+ | 38 | 853 | 300-1600 |
| LB CD19+ | 0.4 | 9 | 200-2100 |
| Cel. CD56 | 7.1 | 159 | 100-1000 |
| CD4/CD8 | 0.71 | 1.5-2.5 | |
Con los antecedentes clínicos del paciente, un patrón de herencia ligado al sexo, el hallazgo de todos los isotipos de inmunoglobulinas disminuidos y de los LB casi ausentes se estableció el diagnóstico de agamaglobulinemia ligada al sexo.
La respuesta exitosa al tratamiento con gamaglobulina IV confirma este diagnóstico. Desde el comienzo de la terapia la frecuencia y severidad de sus infecciones ha disminuido significativamente y la calidad de vida del paciente se ha mejorado de FORMA considerable.