Caso número 2
Paciente de sexo masculino, remitido al grupo IDP a los ocho años y 10 meses de vida con un cuadro de infección recurrente anormal. En orden cronológico sus manifestaciones clínicas se iniciaron entre los tres y cuatro años de edad con bronquitis, OMA y sinusitis por lo cual recibió terapia antibiótica. A los cuatro años se le diagnóstico reflujo gastroesofágico (RGE), que se manejó quirúrgicamente.
Desde los 4 años presentaba cada 15 días episodios de BNM, OMA no supurada y sinusitis, manejadas ambulatoriamente.
A los seis años presentó una neumonía del lóbulo medio que sumada a su sintomatología clínica hizo pensar en tuberculosis (TBC) pulmonar, por lo cual se hospitalizó y se inició tratamiento anti-TBC; seis meses más tarde se sospechó en una reactivación de TBC o en neumonía adquirida en la comunidad.
A los siete años y medio se le practicó lobectomía de pulmón por diagnóstico de síndrome de lóbulo medio; a los ocho años presentó neumonía con edema pulmonar y síndrome sinobronquial. Además de lo anterior el niño presentaba EDA recurrente.
En febrero de 2001 se estableció el diagnóstico de inmunodeficiencia primaria humoral y comenzó a ser tratado con gamaglobulina humana intravenosa.
Este niño es producto del segundo embarazo, durante el cual la madre tuvo control prenatal. El parto fue institucional, sin anoxia perinatal, con una talla y peso al nacer adecuados para la edad gestacional (51 cm y 3300 g). La caída del cordón ocurrió a los 10 días devida.
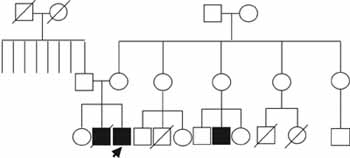
Recibió LACTANCIA materna durante aproximadamente dos años. Ha recibido 5 dosis de polio oral, 3 de DPT, 1 de BCG y 1 de sarampión. Sin antecedentes familiares relevantes con relación a su condición (Figura 4).

Figura 4. Genealogía del caso número dos. El patrón de herencia no puede ser determinado con esta información. El caso índice se señala con una flecha.
Los exámenes realizados desde la sospecha clínica de inmunodeficiencia primaria humoral mostraron niveles de inmunoglobulinas séricas casi ausentes (IgG 70 mg/dl, IgM 14 mg/dl, IgA 25 mg/dl). Luego de la administración de la primera dosis de gamaglobulina, la IgG ascendió a 180 mg/dl y con la segunda dosis la IgG aumentó a 430 mg/dl.
Al llegar al servicio clínico del grupo de Inmunodeficiencias Primarias, se diagnosticó un síndrome de infección recurrente anormal como consecuencia de una inmunodeficiencia humoral. Al examen físico detallado se encontró un niño en buenas condiciones generales, con buen estado nutricional, amígdalas hipotróficas, hipertrofia de cornetes medios en fosa nasal izquierda y a la auscultación en región media de hemitórax derecho disminución del murmullo vesicular; el resto del examen fue completamente normal. Con el fin de descartar la posibilidad de fibrosis quística se le realizó una iontoforesis, que resultó completamente normal. Se analizaron las poblaciones de LT, LB y células NK, resultados que evidenciaron unos LB casi ausentes en sangre periférica (Tabla 3).
Tabla 3.Valores de poblaciones de LT, LB y células NK del caso número dos.
| Descripción | Valor (%) | Células x ml | Valor esperado |
| Leucocitos | 6750 | ||
| Linfocitos | 22 | 1485 | 1700-6900 |
| LT CD3+ | 91.5 | 1359 | 900-4500 |
| LT CD4+ | 59.5 | 884 | 500-2400 |
| LT CD8+ | 28 | 416 | 300-1600 |
| LB CD19+ | 1 | 15 | 200-2100 |
| Cel. CD56 | 13.4 | 299 | 100-1000 |
| CD4/CD8 | 2.12 | 1.5-2.5 | |
Con los antecedentes clínicos del paciente, el hallazgo de los isotipos de inmunoglobulinas disminuidos y de los LB prácticamente ausentes, además de la respuesta exitosa a la terapia con gamaglobulina intravenosa, se diagnosticó una agamaglobulinemia ligada al X. Desde el comienzo de la terapia ha habido una disminución significativa de la frecuencia y severidad de las infecciones, y la calidad de vida del paciente ha mejorado de FORMA considerable.
Caso número 3
Paciente de sexo masculino, remitido al grupo IDP a los 4 años y 5 meses de vida por presentar infecciones recurrentes. Sus manifestaciones se iniciaron a los 6 meses de edad con OMA más una infección estafilocócica en piel, por lo cual fue hospitalizado y tratado con vancomicina. Entre los 9 meses y los 2 años el paciente presentó cuadros gripales frecuentes.
A los 2, 3 y 3 años y medio fue hospitalizado (entre 1 y 3 meses) por presentar episodios de neumonía. A los 4 años y medio sufrió una convulsión por hipoglucemia, por lo que fue hospitalizado durante 25 días.
Este niño es producto del segundo embarazo. El parto fue institucional por cesárea, sin anoxia perinatal, con un peso al nacer de 1700 g. El bajo peso al nacer se debió a que el embarazo fue múltiple (trillizos). La caída del cordón ocurrió a los 20 días de vida.
Recibió LACTANCIA materna durante aproximadamente 2 meses. Su esquema de vacunación ha consistido en una dosis de BCG, 3 de DPT, 4 de polio oral, 3 de hepatitis B, 3 de Haemophilus influenzae, 1 de hepatitis A, 1 de varícela, 1 de SRP, 1 de meningococo, 1 de neumococo y 1 antigripal. No se describen antecedentes familiares relevantes con relación a su condición (Figura 5).

Figura 5. Genealogía del caso número tres. No fue posible determinar el patrón de herencia con la información obtenida. El caso índice se señala con una flecha.
Los exámenes realizados antes del ingreso al servicio clínico del grupo IDP, mostraron que había una reducción marcada de los niveles de inmunoglobulinas séricas. Al llegar al servicio se estableció un diagnóstico de infección recurrente anormal por los datos de la historia clínica y se sospechó de una inmunodeficiencia primaria humoral.
El examen físico detallado reveló un marcado retraso pondoestatural, regular estado general, con palidez generalizada, NUTRICIÓN regular, conjuntivas pálidas, estreches marcada del conducto auditivo externo izquierdo, hipertrofia del cornete medio en fosa nasal derecha, amígdalas hipotróficas, adenopatías cervicales e inguinales pequeñas, en ambos campos pulmonares se auscultó movilización de secreciones y edema marcado de rodilla izquierda.
La evaluación de laboratorio (Tabla 4) evidenció una disminución severa en los LB, una reducción de la fracción gamma en la electroforesis de proteínas y niveles de inmunoglobulinas disminuidos antes de iniciar la terapia con gamaglobulina intra-venosa (IgG 16 mg/dl, IgM 4 mg/dl, IgA 1 mg/dl).
Luego de tres días de haber recibido la primera dosis de gamaglobulina IV los niveles de IgG aumentaron a 584 mg/dl.
Tabla 4. Valores de poblaciones de LT, LB y células NK del caso número tres.
| Descripción | Valor (%) | Células x ml | Valor esperado |
| Leucocitos | 5225 | ||
| Linfocitos | 77 | 4023 | 1700-6900 |
| LT CD3+ | 87.5 | 3520 | 900-4500 |
| LT CD4+ | 38.8 | 1561 | 500-2400 |
| LT CD8+ | 44.2 | 1778 | 300-1600 |
| LB CD19+ | 1 | 40 | 200-2100 |
| Cel. CD56 | 6.4 | 257 | 100-1000 |
| CD4/CD8 |
0.87 |
1.5-2.5 | |
Con los datos presentados en este resumen de historia se estableció el diagnóstico de agamaglobulinemia congénita ligada al X.
Desde el comienzo de la terapia ha habido una disminución significativa de las infecciones y la calidad de vida del paciente se ha mejorado de FORMA considerable.
Discusión
En nuestro medio existe un gran desconocimiento sobre las inmunodeficiencias primarias y secundarias por parte de muchos profesionales de la salud; debido a esto en la mayoría de los casos es difícil hacer un diagnóstico con el tiempo suficiente para que mejore el pronóstico y la calidad de vida de los individuos afectados y en algunos casos inclusive el diagnóstico no se establece.
Dentro de las razones que explican el subregistro de casos y la demora para llegar a un diagnóstico cabe destacar la formación académica recibida por el medico, las políticas de salud de nuestro país, la poca atención a la historia clínica y antecedentes familiares del individuo, el desconocimiento no solo del síndrome de infección recurrente anormal sino también de los grupos que ofrecen un servicio especializado e integral a los individuos afectados por alguna de estas enfermedades y la falta de recursos para realizar pruebas que AYUDEN EN el diagnóstico diferencial de estas entidades.
Nosotros demostramos en este reporte como con una historia clínica bien documentada y unos pocos exámenes de laboratorio se puede llegar a un diagnóstico del síndrome de infección recurrente anormal por una inmunodeficiencia humoral y por último al diagnóstico altamente presuntivo de XLA.
Esperamos que en poco tiempo sea posible realizar el siguiente paso en el estudio de estos pacientes que consiste en una búsqueda en el DNA que codifica para el gen BTK de las mutaciones responsables de este fenotipo, así como a los familiares en quienes se sospeche la enfermedad o el estado de portador.
Casos de agama-globulinemia ligada al X
Pero aunque la mayor parte de los casos de agama-globulinemia ligada al X se asocian a niveles muy bajos o ausentes de inmunoglobulinas séricas, se ha demostrado que entre el 10-20% de los individuos que padecen esta inmunodeficiencia tienen niveles normales o casi normales de inmunoglobulinas (2).
De otro lado no todos los pacientes en quienes se presume XLA tienen mutaciones en el gen de la BTK, se han descrito mutaciones en la cadena pesada de m y en otros componentes del complejo que FORMA el receptor en células pre-B incluyendo ¡5/14.1 que causan un desorden clínicamente idéntico a XLA (2).
En todos los casos presentados, los pacientes se beneficiaron del tratamiento integral logrando un cambio radical en su calidad de vida y la de su familia. Además se redujeron los gastos por parte de las familias y del Estado gracias a la disminución de las frecuentes hospitalizaciones y el uso de fármacos.
En la actualidad los pacientes se encuentran en buen estado sin presentar episodios de infecciones.
En el país se hace necesario crear más centros con capacidad de diagnóstico y manejo de las inmunodeficiencias primarias. Ofreciéndole a los afectados la posibilidad de contar con los mas modernos y eficaces esquemas de tratamiento que den una solución a su dolencia o mejoren su calidad de vida y pronóstico.
Referencias Bibliográficas
- 1. Ochs HD, Smith CI. X-linked agammaglobulinemia. A clinical and molecular analysis. Medicine (Baltimore). 1996; 75: 287-99.
- 2. Minegishi Y, Rohrer J, Conley ME. Recent progress in the diagnosis and the treatment of patients with defects in early B-cell development. Curr Opin Pediatr. 1999; 11: 528-32.
- 3. Hand D Ochs, CI Eduard smith, Jennifer M Puck. Primary Immunodeficiency diseases: a molecular and genetic approach. New York, Oxford University Press. 1999; chapter 22: 263-284.
- 4. García MC, López E, Cambronero R, et al. Diagnóstico molecular de las inmunodeficiencias primarias. Allergol et Immunopathol. 2001; 29: 107-113.
- 5. Abul K Abbas, Andrew H Lichtman, Jordan S Pober. Celular and molecular immunology. Third edition. Saunders company. 1997; chapter 21: 439-444.
- 6. Plebani A, Fischer MB, Meini A, et al. T cell activity and cytokine production in X-linked agammaglobulinemia: implications for vaccination strategies. Int Arch Allergy Immunol. 1997; 114: 90-3.
- 7. Quartier P, Debre M, De Blic J, et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: a retrospective survey of 31 patients. J Pediatr. 1999; 134: 589-96.
- 8. Goddard EA, Beatty DW. X-linked agammaglobulinaemia. S Afr Med J. 1989; 76: 605-7.
- 9. https://www.ncbi.nlm.nih.gov/htbin-post/Omim/getmap?l300300.