
Diana García de Olarte MD.1, Carlos Julio Montoya Guarín, MD., MSc.2, Helí Salgado Vélez MD.3, Pablo Javier Patiño Grajales MD, MSc. DSc4, María Margarita Olivares Gómez, MD5.
1. Pediatra, DSc, Profesor Titular Universidad de Antioquia.
2. Profesor del Centro de Investigaciones Médicas.
3. Pediatra, Inmunólogo, Profesor del Departamento de Microbiología y Parasitología.
4. MSc, DSc, Profesor del Departamento de Microbiología y Parasitología.
5. Programa Detección y Manejo del Síndrome de Infección Recurrente.
Grupo Patogénesis de las Inmunodeficiencias Primarias,
Facultad de Medicina, Universidad de Antioquia.
Las inmunodeficiencias primarias son defectos genéticos que comprometen la función de alguna de las proteínas involucradas en la respuesta inmunológica; por ello, los pacientes presentan una frecuencia anormal de infecciones severas recurrentes, enfermedades autoinmunes, alergias y neoplasias.
Uno de los órganos más comprometidos es la piel, y se ha observado que los diferentes tipos de deficiencia están asociadas con grupos específicos de manifestaciones dermatológicas.
La caracterización oportuna de las alteraciones cutáneas en individuos con sospecha de inmunodeficiencia, es útil a la hora de hacer el diagnóstico fenotípico de la entidad, y en algunos casos, como la enfermedad de injerto contra hospedero en un lactante, sirve para detectar una inmunodeficiencia primaria en ausencia de otras manifestaciones físicas.
Palabras claves: inmunodeficiencias primarias, alteraciones dermatológicas.
Abstract
Primary immunodeficiencies are genetic defects, which can alter the function of those proteins involved in the immune response. Affected individuals have an abnormal frequency of severe and recurrent infections, autoimmune diseases, allergy and cancer.
In patients with immunodeficiency, one of the most frequently affected tissues is the skin, and it is clear that the different types of deficiency are associated with a specific kind of dermatologic manifestations.
Early characterization of the cutaneous alterations in an individual with suspected immunodeficiency is useful to do the phenotypic diagnosis and, in some instances as the graft versus host disease in an infant, to detect a primary immunodeficiency disease when other clinical manifestations are absent.
Key words: primary immunodeficiency, dermatologic alterations.
Introducción
Las inmunodeficiencias primarias (IDP) son trastornos congénitos o heredados por defectos en los genes que codifican alguna de las proteínas involucradas en la respuesta inmunológica (1). De acuerdo con el gen afectado se pueden producir en forma aislada o mixta, diferentes defectos en la respuesta mediada por anticuerpos, los linfocitos T (LT), las células fagocíticas y el sistema del complemento.
Hasta hoy se han definido cerca de cien enfermedades por deficiencia inmunológica, y su clasificación es realizada desde hace varios años por un grupo de investigadores de la Organización Mundial de la Salud (2), y para la región por el Grupo Latinoamericano de Inmunodeficiencias Primarias (LAGID) que tiene un registro de los casos de IDP organizados en seis grupos: deficiencias combinadas, déficit predominante de anticuerpos, deficiencias celulares y de anticuerpos asociadas con otros defectos mayores, inmunodeficiencias asociadas a alteraciones en la función de las células fagocíticas, defectos primarios del sistema fagocítico y defectos congénitos del complemento (3).
El número total de genes en los humanos está próximo a los 100.000. Los comprometidos en el desarrollo y la función de los leucocitos son alrededor de 10.000, y unos 1.000 están relacionados con las IDP, lo cual indica que estas entidades pueden exceder en diez veces el número identificado en la actualidad (1).
La incidencia de las IDP es variada:
Desde la relativamente común deficiencia selectiva de la IgA (1:700 individuos sanos), inmunodeficiencia común variable (1:75.000), inmunodeficiencia severa combinada (1:100.000), hasta la enfermedad granulomatosa crónica (1:200.000). Estos trastornos tienen una relación hombre:mujer de 5:1 en la infancia y de 1:1,4 en los adultos (4).
Las IDP se manifiestan en forma característica a los pocos días de nacido o en la lactancia, pero algunas veces sólo son evidentes en una edad más avanzada. Como consecuencia de estas deficiencias, los individuos presentan en forma anormal infecciones recurrentes severas, enfermedades autoinmunes, alergias y neoplasias, especialmente de origen hematológico.
Las principales puertas de entrada para los diferentes agentes infecciosos son la piel y las mucosas; por esto, los individuos afectados por una IDP presentan enfermedades cutáneas y de los aparatos respiratorio y digestivo.
Desde estos órganos, los microorganismos se diseminan por continuidad o por vía hematógena para causar infecciones en órganos profundos.
Manifestaciones cutáneas de las IDP
Los defectos genéticos en los diferentes componentes de la respuesta inmune se caracterizan por alteraciones moleculares, que inciden en forma directa en la aparición de síntomas y cuadros clínicos, cuya especificidad está directamente relacionada con dichos trastornos.
La piel es el órgano blanco afectado con mayor frecuencia. Las manifestaciones cutáneas tienen un gradiente de severidad que se debe conocer, ya que algunas enfermedades dermatológicas de ocurrencia común pueden ser el primer aviso de una IDP. Las manifestaciones cutáneas asociadas con las IDP son clasificadas por categorías (Tabla 1).
A continuación se presentan breves descripciones de algunos defectos primarios en la respuesta inmune y las características clínicas de las enfermedades dermatológicas asociadas con ellos, en pacientes atendidos durante los últimos 20 años en la Clínica de Inmunodeficiencias Primarias de la Universidad de Antioquia.
(Lea También: Dermatitis Semejante a la Seborreica)
Dermatitis semejante a la atópica
Síndrome de Hiper IgE con Infección Recurrente (SHIEIR)
El SHIEIR es una inmunodeficiencia primaria compleja caracterizada por concentraciones séricas muy elevadas de IgE (> 2000 UI/mL), presencia de abscesos recurrentes en tejidos superficiales y profundos producidos por Staphylococcus aureus. Son frecuentes la dermatitis crónica semejante a la atópica y un síndrome de infección recurrente anormal con desarrollo de neumopatía supurativa y neumatoceles persistentes.
En nuestro medio, en la Clínica de Inmunodeficiencias Primarias de la Universidad de Antioquia, se han caracterizado en los últimos seis años, 11 pacientes con SHIEIR, que corresponden al 17% de las IDP diagnosticadas (4, 5).
Un análisis reciente de 30 pacientes con SHIEIR y de 70 de sus familiares, ha permitido redefinirlo como un desorden multisistémico que afecta el sistema inmune, la piel, la dentición, el esqueleto, y el tejido conectivo.
El defecto molecular responsable no se conoce todavía; no obstante, se hereda como un rasgo autosómico dominante con expresión variable (6).
Los resultados de investigaciones en pacientes con SHIEIR han mostrado una respuesta in vivo anormal de la inmunidad mediada por células y una alteración en la formación de anticuerpos específicos contra antígenos protéicos y polisacáridos (7, 8).
Las causas de estas anomalías, así como los altos niveles séricos de IgE permanecen desconocidas. Existe una incapacidad de los LT para activarse, in vivo e in vitro, cuando son estimulados con antígenos, aunque conservan intacta la respuesta a mitógenos; las pruebas cutáneas de hipersensibilidad retardada son negativas.
Se ha encontrado disminución en la proporción de los LT CD3+/CD45RO+ (fenotipo de LT de memoria), pero número normal de linfocitos CD3+, CD4+ y CD8+. Los granulocitos tienen respuestas quimiotácticas que son variables en el tiempo, la eosinofilia es notable en la sangre y en los tejidos infectados (9, 10).
Tipo de lesión dermatológica |
Inmunodeficiencias primarias |
Dermatitis semejante a la atópica |
Agammaglobulinemia ligada al X; deficiencia IgA; deficiencia IgM; síndrome hiperIgM; inmunodeficiencia común variable; Síndrome de Wiskott Aldrich; Síndrome hiperIgE con infección recurrente; enfermedad granulomatosa crónica |
Dermatitis semejante a la seborreica |
Inmunodeficiencia severa combinada – síndrome de Omenn; ataxia telangiectasia; síndrome de Wiskott Aldrich |
Abscesos cutáneos |
Síndrome hiperIgE con infección recurrente; enfermedad granulomatosa crónica; deficiencia de adhesión de leucocitos tipo I |
Petequias y/o Púrpura |
Primero, Síndrome de Wiskott Aldrich. Segundo, Síndrome de Chediak-Higashi. Tercero, Síndrome de Griscelli |
Telangiectasias mucocutáneas |
Ataxia telangiectasia |
Dilución pigmentaria |
Síndrome de Chediak-Higashi |
Enfermedad de injerto contra hospedero |
Inmunodeficiencia severa combinada; síndrome de DiGeorge; Síndrome de Nezelof |
Granulomas cutáneos |
Enfermedad granulomatosa crónica; ataxia telangiectasia;agammaglobulinemia ligada al X; inmunodeficiencia común variable; inmunodeficiencia severa combinada |
Ulceraciones semejantes al Pyoderma Gangrenoso |
Agammaglobulinemia ligada al X; deficiencia IgA; deficiencia de adhesión de leucocitos tipo I; enfermedad granulomatosa crónica; síndrome hiperIgE con infección recurrente; síndrome de Chediak-HigashiInfecciones cutáneas por Candida albicans inmunodeficiencia severa combinada, síndrome de DiGeorge, candidiasis mucocutánea crónica |
Angioedema |
Angioedema hereditario por deficiencia del C1INH |
Cambios cutáneos semejantes al lupus |
Deficiencia IgA; síndrome hiperIgM; portadores de enfermedadgranulomatosa crónica; deficiencia de primeros componentes del complemento; síndrome de Bloom |
Infecciones virales |
Virus Herpes simplex tipos 1 y 2; varicella zoster, virus EpsteinBarr, citomegalovirus, virus papiloma humano (HPV) asociados con los tipos observados en epidermodisplasia verruciforme (HPV: tipos 5, 8); Molluscum contagiosum, sarampión. |
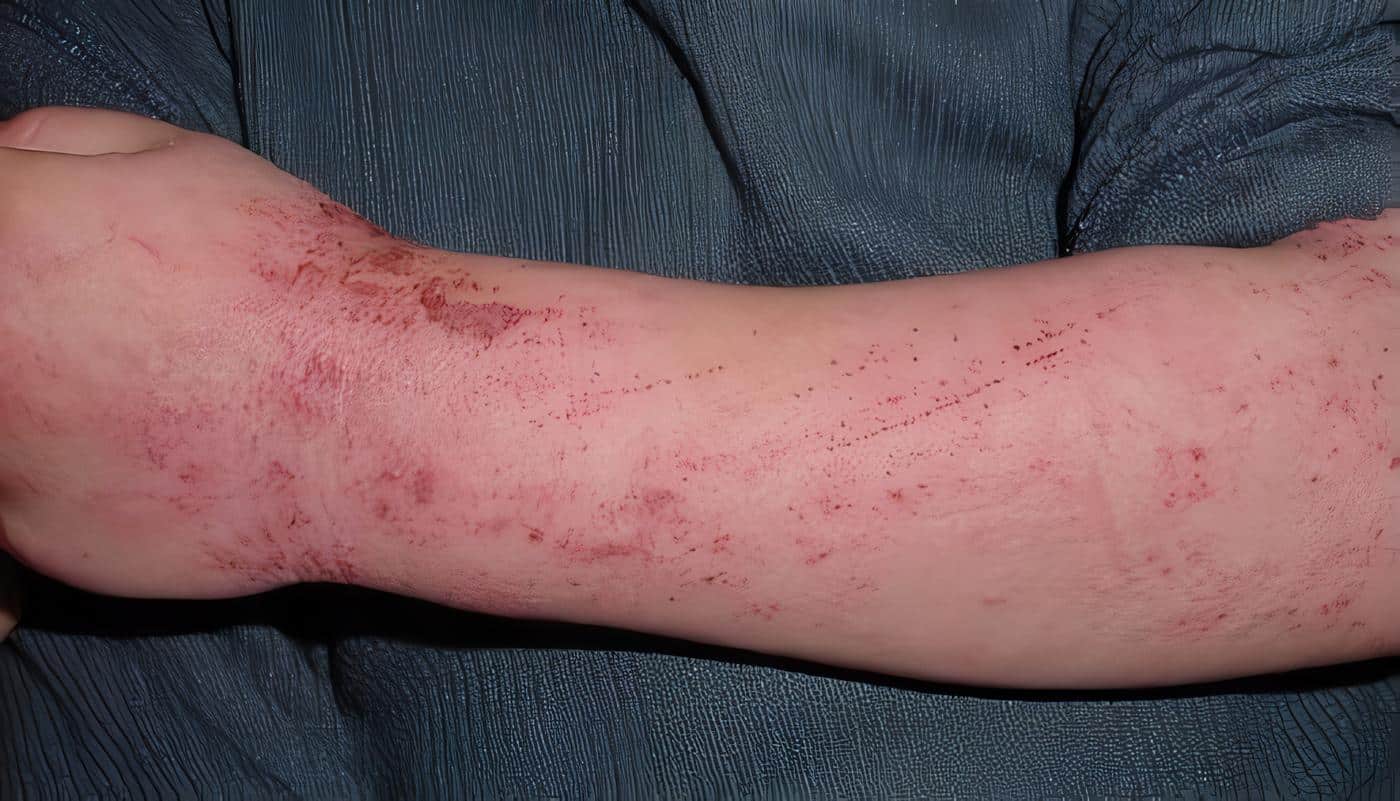
La mayoría de los pacientes presentan infecciones recurrentes desde el primer mes de vida, y la piel es el órgano inicialmente afectado; se encuentran lesiones impetiginizadas que se inician en la cara y el cuero cabelludo y se extienden rápidamente al tronco.
Al mismo tiempo se presentan forúnculos y abscesos en la piel que se denominan “fríos” por la carencia de signos inflamatorios y de toxicidad sistémica, ubicados preferentemente en el cuero cabelludo, la cara y la parte posterior del cuello (Foto 1).

Foto 1. SHIEIR. Absceso “frío” -sin signos inflamatorios- en el cuero cabelludo.
También desde el primer mes de vida hacen su aparición lesiones de dermatitis semejantes a la atópica, de carácter crónico y muy pruriginosa, con una distribución “anti-atópica” que compromete las superficies de extensión (Foto 2). No hay antecedentes personales en el SHIEIR de alergia a alimentos, rinitis o asma.

Foto 2. SHIEIR. Lesiones eritemato pápulo escamosas en mejillas y mentón, semejantes a las atópicas.
Con el paso de los años, los pacientes con SHIEIR presentan variaciones en el desarrollo cráneo-facial, se hace evidente una dismorfia facial caracterizada por aumento de la circunferencia de la cabeza, hueso frontal prominente, puente nasal amplio, aumento de la distancia entre las alas de la nariz, engrosamiento de la punta de la nariz y de los labios y prognatismo moderado (11) (Foto 3).

Foto 3. Facies dismórfica característica del SHIEIR. Asimetría facial, circunferencia alargada de la cabeza, frente prominente, puente nasal amplio, aumento de la distancia interalar, engrosamiento de la nariz y de los labios. Cambios iniciales de distrofia ungueal y de falanges distales de los dedos.
Otras alteraciones dermatológicas son la candidiasis recurrente de piel y mucosas, y en la cara la piel es áspera y con cicatrices pequeñas circulares y puntiformes, similares a las resultantes de las secuelas de la varicela o el acné (Foto 3).





hola me interesa saber la referencia bibliográfica de este articulo ya que lo necesito citar en un nuevo articulo muchas gracias
Buenas tardes Cesar, esta entrada hace parte de la Revista de Inmunoalergia Vol 09 nº 4 https://encolombia.com/medicina/revistas-medicas/alergia/vol-940/