En los pacientes con amenorrea primaria, el trastorno se encuentra con bastante frecuencia localizado en las gónadas, que pueden ser disgenéticas, como en los casos típicos de Turner, en las disgenesias gonadales mixtas o atípicas (donde hay talla baja con otras malformaciones, mosaicismo con presencia de un Y, genitales externos hipoplásicos femeninos o ambiguos, genitales internos con estructuras masculinas y femeninas -cintillas fibrosas y testículo disgenético-), o en las disgenesias gonadales puras (6, 7, 8, 9l. Se pueden también observar casos de pseudohermafroditismo masculino como los testículos feminizantes, la regresión testicular (agonadismol o defectos enzimáticos (8,10).
Finalmente los raros casos de tumores ováricos productores de andrógenos pueden originar también una amenorrea primaria.
Síndrome de Turner.
Desde que Turner informó en 1938 los primeros siete casos, esta entidad se ha convertido en una de las causas más comunes de amenorrea primaria.
Presentan variadas malformaciones como corta estatura y cuello alado, anormalidades de la cabeza, ojos, oídos, como hipertelorismo ocular, epicantus, implantación baja de las orejas, alteraciones en la forma del paladar; deformidades torácicas e hipertelorismo mamario, anomalías cardiovasculares como la coartación de la aorta, hipertensión y telangiectasia intestinal.
Obviamente hay hipoplasia de genitales externos e internos -infantilismo sexual-, y amenorrea primaria. También hay manifestaciones esqueléticas con retardo en la maduración ósea, cúbitus valgus, 40. metacarpiano corto, osteoporosis, edema de manos y pies que se pueden observar en la infancia, cutis laxa, queloides e hipoplasia de las uñas.
Muy ocasionalmente se pueden advertir déficit intelectuales6.
Entre 1968 y 1978 tuvimos la oportunidad de seguir 11 casos de síndrome de Turner en la consulta de endocrinología del Hospital San Ignacio. Desde esa fecha, tenemos una incidencia aproximada de un caso nuevo al año (Fig. 5).
E180% de los pacientes tienen el típico cariotipo 45 XO, pero pueden existir también mosaicismos.
Las gonadotrofinas LH y FSH están característicamente elevadas, con niveles no detectables de estradiol; hay gónadas rudimentarias e hipoplasia uterina a la ecografía y laparoscopia. Hay buena respuesta al tratamiento de suplencia con estrógenos y progestágenos.
Testículos Feminízantes.
Una interesante variedad de pseudohermafroditismo masculino es esta enfermedad familiar que se transmite, bien con un gen recesivo ligado al sexo o como un gen autosómico dominante limitado a los hombres.
Las gónadas -de tipo masculino-, que se encuentran en el abdomen, han sido descritas como testículos fetales, inmaduros o criptorquídicos.
La producción de andrógenos y estrógenos es normal en estas pacientes pero los tejidos son refractarios a la acción androgénica; el fenotipo es femenino mientras que el cariotipo es masculino.
En la década de los 70, Ospina y Medina, estudiaron en el Hospital San Ignacio a tres hermanas con este SíndromelO, de 16, 18 Y 20 años; presentaban características similares, con talla y peso adecuados para la edad, con senos bien desarrollados, vello axilar ausente, vello púbico ausente en la menor, muy escaso en las dos mayores; genitales externos femeninos con vagina ciega de 4, 6 y 7 cmts. de longitud, respectivamente; gónadas abdominales con ausencia de útero y trompas; cariotipo 46 XY en las tres yun valor estrogénico medio de 72.7,52.3 y 53. 7, respectivamente.
El estudio histológico de las gónadas -tanto a la microscopia de luz como electrónica -, descrito en detalle por Ospina y Medina, mostró ausencia de espermatogénesis, células de Sertoli que ocupaban casi toda la luz de los túbulos seminíferos y un espacio intersticial formado por células de Leydig y tejido conectivo.
 Fig. 5. Paciente con Síndrome de ‘fumer. Obsérvese la corta estatura (1.21 mts. para 12 años), hipertelorismo ocular y mamario, cuello corto, implantación baja de las orejas, epicantus, cúbitus va1gus e infantilismo sexual.
Fig. 5. Paciente con Síndrome de ‘fumer. Obsérvese la corta estatura (1.21 mts. para 12 años), hipertelorismo ocular y mamario, cuello corto, implantación baja de las orejas, epicantus, cúbitus va1gus e infantilismo sexual.
Después de la gonadectomia – realizada para evitar una degeneración maligna-, las pacientes presentaron oleadas de calor, cansancio y cefalea, por lo que se trataron con terapia de suplencia a base de estrógenos conjugados.
Una de las pacientes recibió metil-testosterona como única terapia, pero al cabo de seis meses no se apreció ninguna respuesta ni clínica, ni en la citología vaginal, demostrándose la refractoriedad a los andrógenos.
Estas tres hermanas pertenecen al grupo de pacientes con síndrome completo; cuando hay algunos signos de virilización como clitoromegalia y algo de vello axilar, se considera incompleto.
Agonadismo XY.
Una paciente de 17 años consultó por amenorrea primaria. Al examen, tenía una estatura de 1.58 mts.; 47.5 kg. de peso; fenotipo femenino, infantilismo sexual, había escaso vello púbico, clítoris y uretra normales; la vagina estaba presente.
No había desarrollo mamario (Fig. 6 y 7); los exámenes de laboratorio mostraron una LH de 59.7 mUllml; Estradiol no detectable, valor estrogénico medio de cero a la citologia; Testasterona total de 0.15 mg/ml.; 17-hidroxiprogesterona, 0.1 mg/ml.; DHEA 71.8 ug/dl.; TSH 3.8 uUllmI.; T410.4 ug/dl.; cariotipo 46 XY (Fig. 8). Se practicó laparotomia, encontrándose todas las estructuras de Müller (útero, trompas) completas. Había ausencia de gónadas en la cavidad pélvica y en los conductos inguinales (Fig. 9).
Se examinó tejido del sitio donde deberían estar las gónadas, lo que sólo mostró tejido fibroso y muscular. Se inició terapia de suplencia, con estrógenos y progestágenos, por lo que presentó desarrollo de los senos y ciclos regulares. Este síndrome de fenotipo femenino con genitales internos, cariotipo masculino, hipergonadotropinismo (con valores de menopausia) y ausencia de estrógenos, con niveles de andrógenos en el límite bajo de lo normal, nos llevó a hacer el diagnóstico de un síndrome de regresión testicular o agonadismo XY.
Fig. 6. Paciente con agonadismo XY. Hay fenotipo femenino con ausencia de telarquia y de vello axilar.
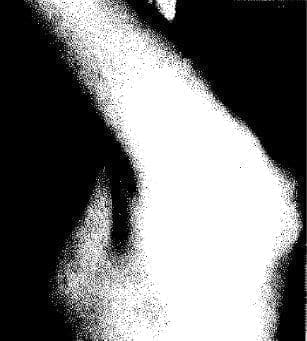
Fig. 7. Genitales externos femeninos infantiles en la paciente con síndrome de regresión testicular (agonadismo XY). Hay escaso vello púbico, clítoris de tamaño normal, vagina presente.
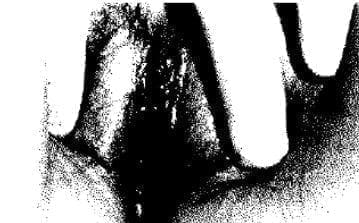
Fig. 8. Cariotipo XY en el mismo caso de regresión testicular y fenotipo femenino.
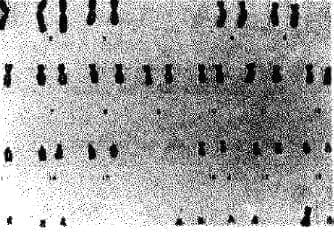
Fig. 9. Obsérvese útero y trompas a la laparotomia.
Falla ovárica prematura.
Una paciente de 28 años consultó porque a los 15 años tuvo dos episodios menstruales y luego entró en amenorrea.
Al examen se encontró una paciente de fenotipo femenino, con estatura de 1.65 mts. y 63 kilos de peso; desarrollo de caracteres sexuales secundarios estado II a III según la clasificación de Tanner, con genitales externos femeninos, útero hipoplásico, ovarios no palpables. Había aumento de vello en miembros inferiores.
Los exámenes de laboratorio mostraron una FSH de 94.1 mUI/mI.; LH de 27.3 mUIImI.; Estradiolde 35 pg/mI.; Progesterona de 0.1 mg/mI. y un valor estrogénico medio de 52.5, o sea, una deficiencia de hormonas sexuales con hipergonadotropinismo; cario tipo de 46XX. Las pruebas de función tiroidea mostraron una T4 de 6.1 ug/dI., índice de T4libre de 2.6 y TSH de 13.6 uUI/mI.
El hipo tiroidismo subclínico se confirmó con una prueba de TRH que dio una respuesta magnificada con TSH (en uUI/mI.) basal de 11.6, 87.5 alos 30 minutos, y 75.8 a la hora. Los exámenes de química sanguínea fueron normales, excepto la colesterolemia que estaba en 282.1 mg/dI.
El calcio, el fósforo y la PTH fueron normales; la radiografía de silla turca fue normal; el carpograma mostró una edad ósea de 17 años y la radiografía de columna dorso-lumbar mostró un proceso osteo-condrósico tipo Schwermann (Fig. 10).
La ecografía mostró un útero pequeño con ovarios elongados y la laparoscopia confirmó útero hipoplásico con ovarios atrésicos (Figs. 11 y 12).
Se hicieron los diagnósticos de falla ovárica prematura de tipo afolicular asociada a cariotipo normal, hipo tiroidismo primario subclínico y epifisítis vertebral tipo enfermedad de Schwermann. Se realizó un tratamiento de suplencia con estrógenos conjugados y medroxiprogesterona, con buenos resultados; aunque esta paciente representa una amenorrea secundaria, la incluimos por lo prematuro de la desaparición de sus reglas.
Fig.l0. Proceso osteocondrósico vertebral tipo Schwermann (en una paciente con falla ovárica prematura).

Fig. 11. La pinza de Rochester muestra la gónada atrésica a la laparoscopía.

Fig. 12. La histología ovárica muestra ausencia de folículos en la paciente con falla ovárica prematura.

Trastornos Suprarrenales y Tiroideos
La hiperplasia suprarrenal congénita, una entidad de transmisión genética y susceptibilidad ligada a los antígenos HLA en el brazo corto del cromosoma 6, tiene una incidencia variable, según la región del mundo donde se hagan los estudios; en los E stados Unidos, por ejemplo, la incidencia es de uno en 50.000, aproximadamente.
Como el diagnóstico se hace al nacimiento o en una edad temprana -también se puede hacer “in útero” -, el tratamiento adecuado en las mujeres afectadas permite la aparición de la menarquía. En los hombres hay macrogenitosomia (pseudopubertad) precoz. Si la mujer está en edad puberal pero no se ha hecho el diagnóstico o se ha suspendido el tratamiento, se observará una amenorrea primaria, con pseudohermafroditismo femenino por virilización.
Hasta 1978 se habían estudiado en el Hospital San Ignacio siete pacientes (cinco mujeres y dos hombres) con hiperplasia suprarrenal por deficiencia de 21-hidroxilasa, parcial en seis y con bloqueo completo en unoll.
Posteriormente se han observado algunos casos más, tanto de la variedad congénita como de la llamada “de aparición tardía”, con manifestaciones de hirsutismo, oligomenorrea o amenorrea secundaria.
Los niveles de andrógenos están elevados, estando también incrementados en forma característica los niveles serios de 17-hidroxi-progesteronay los urinarios de pregnanetriol.
Los tumores ováricos productores de andrógenos -que rara vez se observan-, pueden cursar con amenorrea primaria. Igualmente, puede ocurrir en las pacientes con hipotiroidismo juvenil (o con Enfermedad de Graves), en quienes no se ha iniciado tratamiento para su enfermedad de base.
Hiperplasia suprarrenal congénita y amenorrea primaria.
En 1983 se recibió en el Servicio de Medicina Interna del Hospital San Ignacio a una paciente de 22 años, referida para estudio de artralgias. Presentaba amenorrea primaria y antecedentes de hirsutismo y acné desde temprana edad.
Al examen se encontró una paciente con talla corta, hábito androide, acné facial, senos hipotróficos con escaso tejido mamario derecho, vello púbico androide, genitales externos sin clítoris (aunque se palpaba cordón fibroso desde pubis hasta introito vaginal).
No había desarrollo de labios mayores ni menores, había un esbozo de introito vaginal sin observarse meato uretral y al tacto rectal se palpaba masa pequeña, móvil, paramediana derecha que parecía corresponder a útero pequeño. Los anexos estaban libres (Fig. 13).
Fig.13. Obsérvese hirsutismo en las piernas y genitales externos (sin clítoris) en una paciente con hiperplasia suprarrenal congénita.

Al revisar la historia se encontró que la paciente había sido estudiada 15 años antes por nosotros, cuando a los siete años consultó por pubarquía, hipertrofia clotoridiana desde el nacimiento, con erecciones en los últimos 18 meses.
Al examen se encontró una paciente de 1.32 de estatura, hipertrofia del clítoris (de tres centímetros con capuchón redundantes), vulva normal e introito con vagina rudimentaria, vello púbico escaso. La edad ósea estaba en once años, los 17-cetoesteroides urinarios estaban en 10.0 mgs. en 24 horas (normal, menos de 2.5), 17-hidroxicorticoesteroides urinarios de 1.3 mg./24 horas, pregnanetriol urinarío en 12.3 mg.l24 horas, con buena supresión postbetametasona de los 17-eetoesteroides que descendieron a 1.3 rng.l 24 horas.
Se le hizo el diagnóstico de hiperplasia suprarrenal congénita y se le instauró el tratamiento supresivo (-practicándose además una clitoridectomía-), pero la madre no le dio el tratamiento ni le explicó a la hija qué tipo de enfermedad tenía.
Durante 15 años no volvió a control.
En la admisión posterior que mencionamos se corroboró el diagnóstico pues presentaba una testosterona elevada de 7.8 nMol/L. y 17-hidroxi-progesterona igualmente elevada de más de 12 nMol/L., cifras que descendieron post-supresión a 1.0 y 1.36, respectivamente.
La laparoscopia mostró genitales internos normales infantiles y la genitografía mostró que la vagina y uretra desembocaban en orificio común. Se practicó vaginoplastia y se inició terapia supresiva con corticoides y suplementarían inicial con estro-progestágenos, con lo que la paciente inició sus menstruaciones, empezaron a aparecer los caracteres sexuales femeninos secundarios y a reducirse el hirsutismo.




