
Los errores fetales en el desarrollo gonadoductal y genital son también causa de ausencia de menarquía; entre estos están los casos de Síndrome de Rokitansky-Küster-Hauser (ausencia de útero), himen imperforado, sinequias endometriales, etc.
Queremos presentar aquí dos casos de Síndrome de Rokitansky, uno clásico y otro con atresia ovárica asociada.
Síndrome de Rokitansky y Atresia Ovárica.
Una paciente de 18 años consultó por amenorrea primaria.
Al examen la estatura era de 1.66 mts., 47 kilos de peso, fenotipo femenino e infantilismo sexual (Fig.14). El laboratorio mostró una FSH de 108 mU I/ml., LH de 53 108 mU I/ml., 17-beta-estradiol de 12 pg/ml., valor estrogénico medio de 50, carpograma con una edad ósea de 12 años, cariotipo 46XX, (Fig. 15); ecografía pélvica con ausencia de genitales internos femeninos lo que se comprobó a la laparoscopia, observándose además una excrecencía de 0.2 mts. de diámetro en repliegue vesical derecho que resultó ser tejido ovárico atré· sico.
Se hicieron entonces los diagnósticos de Rokitansky- Küster-Hauser asociado a ovario ectópico rudimentario e hipogonadismo secundario a falla ovárica asociada a cariotipo normal. Se inició tratamiento de suplencia, con buen desarrollo de los senos (Fig. 16).
Síndrome de Rokitansky Clásico.
Paciente de 23 años que consultó por amenorrea primaria.
Al examen se encontró una paciente con estatura de 1.60, peso de 51 kilos y un desarrollo normal de caracteres sexuales secundarios (Fig. 17). Presentaba una vagina ciega de 2 cmts. de profundidad, con genitales internos no palpables. La ecografía pélvica mostró ausencia de útero y trompas con gónadas presentes.
Con el diagnóstico de Síndrome de Rokitansky- Küster-Hauser, se practicó una vaginoplastia (Fig. 18).
 Fig. 14. Obsérvese la falta de vello axilar y de desarrollo de los senos en una paciente con agenesia uterina -Síndrome de Rokitansky-, y atresia ovárica.
Fig. 14. Obsérvese la falta de vello axilar y de desarrollo de los senos en una paciente con agenesia uterina -Síndrome de Rokitansky-, y atresia ovárica.

Fig. 15. Cariotipo XX (cromosomas fluorescentes) en la paciente con agenesia uterina y atrofia ovárica.

Fig. 16. La misma paciente muestra desarrollo de los senos con el tratamiento estrogénico de suplencia.

Fig. 17. Paciente con Síndrome de Rokitansky, caracteres sexuales secundarios normales.
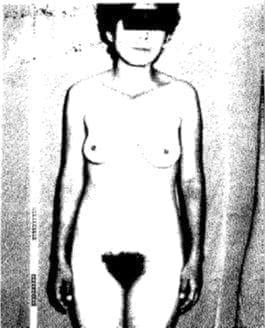
Fig. 18. Se practicó vaginoplastia a esta paciente.

Discusión
En esta revisión general del problema de las amenorreas primarias, el objeto principal es informar el enfoque multidisciplinario que le damos en el Hospital San Ignacio, ilustrando la descripción con alguna casuística interesante.
La amenorrea primaria es sólo el síntoma principal, el cual motiva al paciente a consultar.
Múltiples son, sin embargo, los problemas de salud que estos pacientes presentan; el infantilismo genital, por ejemplo, tiene implicaciones psicológicas y sociales en relación con la familia o con personas de su misma edad, además de las sexuales propiamente dichas, por su incapacidad física de formar pareja sin un tratamiento adecuado y su infertilidad.
El hipoestrogenismo lleva a la osteoporosis, las gónadas masculinas (o sus restos) intraabdominales tienen un potencial carcinogénico y la presencia de hiperandrogenismo traumatiza al paciente; los tumores intraselares o hipotalárnicos pueden además tener repercusiones neurooftalmológicas, mientras que algunos trastornos hereditarios ameritan el estudio genético de la familia.
En nuestra experiencia lo que más comúnmente vemos es el Turner, que usualmente presenta los estigmas clásicos que ya hemos descrito6; las anormalidades anatómicas no son raras, como vemos, por los dos casos presentados de agenesia uterina.
Los microprolactinomas son básicamente un problema de infertilidad con amenorrea secundaria y galactorrea; es pues, una enfermedad benigna Los macroprolactinomas (en hombres o en mujeres) pueden además tener complicaciones en las vias ópticas y algunas otras alteraciones neurológicas.
No es frecuente que éstos causen amenorrea primaria, pero si ocurre, se observa pubarquia y adrenarquia como en la paciente que aqui presentamos.
El advenimiento de los agonistas de la dopamina, tipo bromocriptina, han facilitado el tratamiento, tanto de los micro como de los macroprolactinomas.
Un estudio de investigadores americanos, canadienses y británicos:
Sobre 16 mujeres y 11 hombres, entre 29 y 43 años, con macroprolactinoma (dos de los pacientes ya habian sido operados), con prolactinemias de más de 150 nglml., y con evidencia radiológica de extensión extraselar, fueron tratados por seis meses o más con dosis de bromocriptina que variaron entre 7.5 y 20 mg. diarios.
Todos los pacientes tuvieron disminución en la prolactina y 181a normalizaron; todos los tumores disminuyeron de tamaño y la mitad se aminoró en más del 50%; las manifestaciones clinicas (trastornos visuales, galactorrea, amenorrea, impotencia y pérdida de la libido) mejoraron o desaparecieron en la mayoría de los pacientes.
Sin embargo, se observó re-expansión del tumor en tres de cuatro pacientes, en quienes se suspendió el tratamiento después de un año, lo que confirma que el manejo médico de los prolactinomas, aunque es de elección como tratamiento inicial, debe usarse por largos periodos de tiempo, si no de por vida, particularmente en macroadenomas; otros abogan por la cirugía transesfenoidal5.
El Síndrome de Kallmann o hipogonadismo hipogonadotrópico asociado a anosmia o hiposmia, es una entidad de ocurrencia pocom común. Lieblich y Cols., del Instituto Nacional de la Salud de los Estados Unidos, estudiaron 23 pacientes (9 mujeres y 14 hombresl pertenecientes a 18 familias; dentro de ellos (en 7 familias) hubo varias personas más afectadas, cinco anósmicas pero eugonadales y dos auósmicas pero con hipogonadismo hipogonadotrópico.
El único déficit endocrino clínico en estos pacientes fue el relacitnado con FSH y LH que estaban bajos (a pesar de niveles subnormales de testosterona o estradiol) y su mala respuesta al Clomifeno, con buena respuesta a la gonadotrofina coriónica en los hombres tratados.
Las respuestas a la gonadorrelina (LH -RH) fueron heterogéneas y hubo pacientes con normalidades sutiles en la función hipotálamo-hipofisiaria.
Otras manifestaciones fenotípicas que se presentaron en algunos de estos casos (mas no en el nuestro), fueron obesidad, diabetes, sordera neurosensorial, criptorquidia, ginecomastia, hendiduras labiales o del paladar, osteopenia, y otras manifestaciones de naturaleza genética.
Las manifestaciones más importantes de este Síndrome parece heredarse algunas veces de una manera autosómica recesiva. En los pocos pacientes de la literatura en quienes se ha hecho autopsia, se ha observado agenesia de los bulbos olfatorios junto con “hipoplasia global” del hipotálamo4.
Overzier describió una serie de casos de agonadismo verdadero que corresponde a individuos genéticamente XY, de aspecto habitualmente femenino, estatura normal, usualmente sin anomalías somáticas, ausencia de desarrollo mamario y escaso pelo axilar y pubiano.
No presentan útero ni gónadas, no existe seno urogenital ni vagina y la uretra se abre al exterior en la base del clítoris.
Una de nuestras pacientes ha sido clasificada como agonadismo, en quien se encontró fenotipo femenino, cariotipo XY, ausencia de gónadas a la laparotomía pero con estructuras de Müller presentes, con un hipogonadismo hipergonadotrópico.
Sus genitales externos mostraban escaso vello púbico y vagina; había varias diferencias con los casos de Overzier tales como clítoris normal en nuestro caso (vs. ligera hipertrofia), labios mayores normales (vs. hipoplásicos y fusionados en el rafé medio).
El Síndrome de Overzier se puede explicar por una diferenciación fugaz y temporal del testículo fetal que es suficiente para inhibir el desarrollo de los conductos de Wolff8, en otras palabras, se produce factor inhibitorio de Müller, inhibiéndose la formación de útero, anexos y 2/3 superiores de vagina. Posteriormente se atrofia la gónada masculina, no se produce la testosterona fetal y no se desarrollan las estructuras dependientes de Wolfí.
Nuestro caso ni siquiera alcanzó a producir factor inhibitorio de Müller (por lo que tiene estas estructuras), de manera que es un agonadismo XY más anterior que los descritos por Overzier.
Nuestra paciente con falla ovárica prematura podría corresponder a una variedad incompleta de disgenesia gonadal pura XX o en su defecto, a un Síndrome poliglandular autoinmune (PG A); menos de cien casos se han informado del primer grupo, en los cuales hay un cariotipo 46 XX, que en los casos familiares es de herencia recesiva autosómica (en un porcentaje asociado a sordera neurosensorial), genitales externos e internos femeninos con bien cintilla (atresia) ovárica y ovario hipoplásico, bien con ovario hipoplásico de ambos lados, estatura normal sin estigmas de Turner, pubertad incompleta, falla ovárica prematura y estradiol plasmático disminuido o normal9.
Nuestra paciente debió tener unos pocos folículos que le permitieron tener dos reglas. Luego el ovario se volvió atrésico.
La posibilidad de una PGA Tipo III en la cual hay enfermedad tiroidea autoinmune con alguna otra enfermedad autoinmune (v.gr. falla ovárica primaria) pero sin enfermedad de Addison12, la consideramos por la asociación en nuestra paciente de hipo tiroidismo subclínico y falla ovárica prematura, pero no es posible comprobarlo ya que no determinamos anticuerpos en esta paciente.
Por último, presentamos un par de casos con agenesia uterina (Síndrome de Rokitansky), uno de ellos con atresia ovárica asociada; otras anomalías anatómicas además de ésta que pueden cursar con amenorrea primaria, son el himen imperforado y las sinequias endometriales13.
Para finalizar, queremos comentar que después del examen físico, ayuda mucho la valoración endocrina en la clasificación de estas pacientes.
Un hipogonadismo sugiere problema ovárico si hay FSH y LH altas o hipotálamo-hipofisiario si las gonadotropinas están baj as. Un eje normal podría sugerir anormalidades anatómicas mientras que un hiperandrogenismo, alteraciones en la T4, podrían sugerir causas endocrinas extraováricas (suprarrenales o tiroideas) de amenorrea primaria.
Posteriormente se hacen necesarios los estudios genéticos y los imagenológicos, incluyendo también las exploraciones visuales directas.
Agradecimientos
A la Unidad de Genética Clínica y al Departamento de Radiología del Hospital San Ignacio, por los estudios realizados a nuestros pacientes.
Bibliografia
- 1. ROSS. GT. Vande Wiele RL. The ovaries. “Textbook of Endocrinology”. R.H. Williams (Editor). 6a. Ed ..WB Saunders, Philadelphia. 1981: 355-399.
- 2. JUB IZ, W. Endocrinología Clínica. Manual Modemo. México. 1981:297-321.
- 3. MORENO-ESCALLON. B. Endocrinología del ciclo menstrual humano. Univ. Med .. 1983. 25: 179-192.
- 4. LIEBLICH, JM, Rogel AD. White BJ, Rosen SW. Syndrome of anosmia with hypogonadotropic hypogonadism (Kallmann’s Syndromei. Clínical and Laboratory Studies in 23 cases. Am J Med. 1982. 73: 506-519.
- 5. MOLICH. ME, Elton RL, y Cols del grupo de estudio de bromocriptina: Bromocritine an primary Therapy for prolactin-secreting macroadenomas, results of a prospective multicenter study. J. Clín Endocrinol Metab, 1985.60: 698-707.
- 6. DANOWSKI, TS. Clinical Endocrinology, Williams & Wilkins Co., Baltimore, 1962. Vol. I, Cap. 30, Gonadal Dysgenesis,: 385-398.
- 7. FIGUEROA, P. Ovarios; en “Endocrinología Clinica”. JP. Salvaneschi (Editorl, Ed. El Ateneo, Buenos Aires, 1984.: 169-193.
- 8. MARTINEZ-ARANTES E. Dimorfismos Sexuales; en “Endocrinología Clinica”, JP. Salvaneschi (Editor), Ed. El Ateneo, Buenos Aires, 1984.: 207-33.
- 9. GRUMBACH, MM, Conte FA. Disorders of sex differentiation. “Textbook of Endocrinology”, RH Williams (Editor), 6′ Ed., WB Saunders, Philadelphia, 1981.: 423-514.
- 10. 0SPINA, JE, Medina J. Uitraestructura del Síndrome de Feminización Testicular. Rev. Soc. Col. Endoc., 1978. 11:75-82.
- 11. NUÑEZ, F., Jácome A. Hiperplasia Suprarrenal Congénita, Valoración Hormonal y Morfológíca de Siete Casos. Rev. Soco Col. Endocrinol., 1978. 11:65-74.
- 12. NEUFELD, M., Maclaren NK, Blizzard RM. Two Types of Autoinmune Addison’s Disease Associated with different Poliglandular Autoinmune (PGA) Syndromes. Medicine, 1981. 60:355-362.
- 13. MEDINA, J. Otero E., Mendoza C., Ospina JE, Ardila MC. Amenorrea Primaria, hallazgos clínicos y citogenéticos en quince pacientes. Rev. Col. Obsto Ginecol., 1972. 23:307-317.




