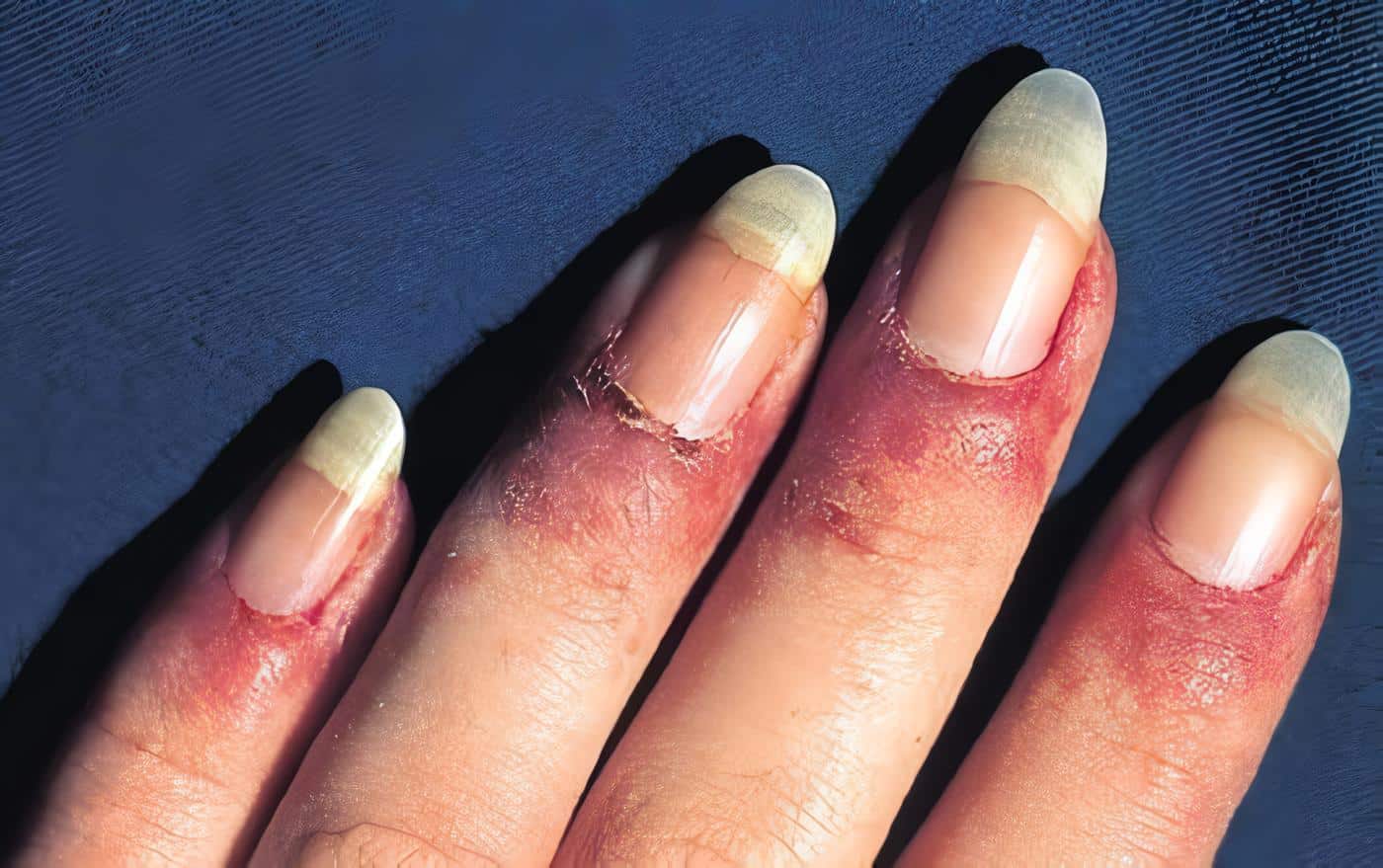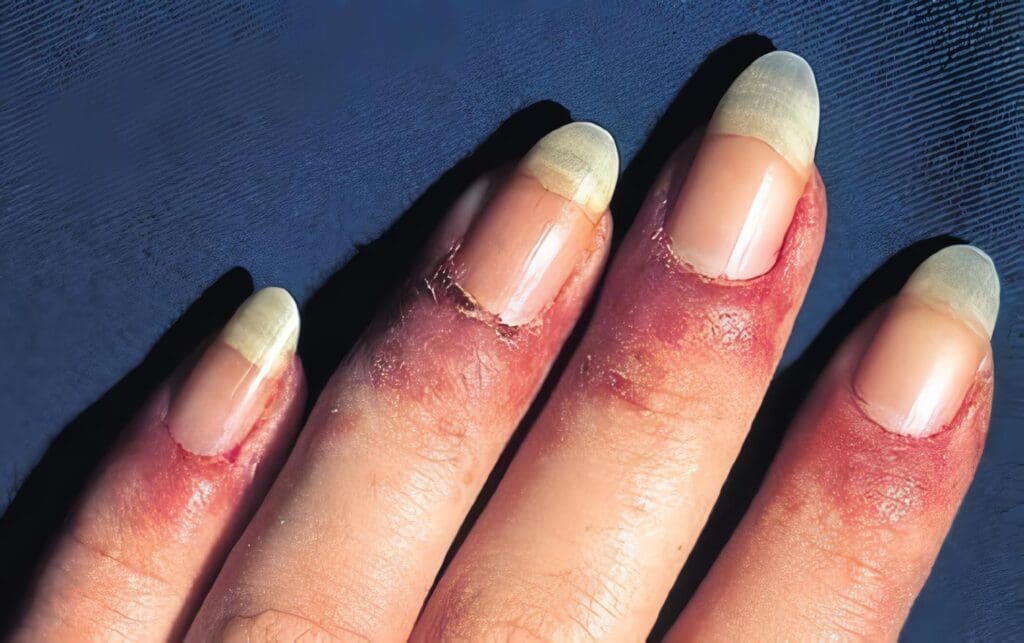
Las crioinmunoglobulinemias asociadas a vasculitis se caracteriza por el depósito de crioglobulinas mixtas en la pared de los vasos (arteriolas capilares y vénulas) ocasionando vasculitis de tipo leucitoclástico.
En 1966, Meltzer y col describieron un síndrome caracterizado por púrpura, artralgias, artritis, síntomas constitucionales fiebre, glomerulonefritis, compromiso abdominal y hepático y depósito de crioglobulinas mixtas en los tejidos inicialmente se denominó esencial por la ausencia en ese momento de causas infecciosas, autoinmunes y/o enfermedades linfo-proliferativas. Las crioglobulinas pueden ser de tipo policlonal IgG o monoclonal o policlonal de tipo IgM y con actividad o no de factor reumatoideo que pueden ser ocasionalmente IgG o IgA.
La vasculitis leucocitoclástica es la manifestación clínica predominante, pero en estudios de autopsias se puede encontrar arteritis visceral en algunos pacientes, y disminución del complemento sérico, con depósitos de IgM, IgG y complemento en las paredes de los vasos y en los glomérulos que explica el papel de los complejos inmunes está relacionado en la patogénesis. Ferry y col en la revista de lupus de 1998 menciona la asociación de crioglobulinemia con hepatitis B y especialmente la C, cirrosis, carcinoma hepatocelular, linfoma no Hodgkin, fibrosis pulmonar, enfermedades linfoproliferativas, tirioditis autoinmunes, hepatitis autoinmune, porfiria cutánea, tarda, glomerulonefritis membrano-proliferativa, lupus y síndrome de Sjögren primario.
Por las asociaciones antes mencionadas esta enfermedad tiene ciertas características peculiares como son la proliferación de linfocitos con producción de autoanticuerpos y complejos inmunes ocasionando un polimorfismo clínico que representa una mezcla de autoinmunidad y linfoproliferación celular, según Brouet y col las crioglobulinas se clasifican en tres subgrupos: crioglobulina monoclonal tipo I, crioglobulinemia tipo II y III caracterizadas por IgG policlonal y IgM monoclonal o policlonal respectivamente. La IgM es un autoanticuerpo con actividad de factor reumatoide.
Las crioglobulinemias tipo I:
Se encuentran principalmente en pacientes con enfermedades linfoproliferativas tales como mieloma múltiple, macroglobulinemia de Waldenstrom, crónicas o enfermedades mediadas por complejos inmunes.
El cuadro clínico lo tomamos de la publicación Ferry y col sobre 150 pacientes, que es una de las series médicas más importantes sobre el tema.
Consideramos que debemos observar bien estos pacientes porque generalmente se asocian a otras patologías o a condiciones co-morbidas que las hace en muchos casos ser refractarias.
De acuerdo a los tipos de crioinmunoglobulinemia se pueden presentar algunos casos refractarios. Ejemplo: En los tipos I y II se caracterizan por oclusiones vasculares con ACV y la necrosis de piel y de extremidades. En algunos casos de crioinmunoglobulinemia tipo II pueden evolucionar a linfoma no Hodgkin. En la de tipo III se presentan casos con vasculitis leucocitoclástica que cursan con muchos episodios de recidiva y en ocasiones su manejo se hace bastante difícil.
Tabla. Crioinmunoglobulinemias. Datos clínicos – epidemiológicos sobre 50 pacientes

Tratamiento
Cuando la enfermedad tiene un curso leve tales como púrpura palpable y artralgias se utilizan esteroides en dosis bajas a moderadas (15 a 30 mg/día), y anti-inflamatorios. Si existe un compromiso visceral, especialmente glomerulonefritis se requiere el tratamiento a base de corticoides combinado con inmunosupresores como la ciclofosfamida. Si ha utilizado la plasmaféresis en algunos casos graves. Pero no se puede sacar conclusiones al respecto. Si el paciente tiene una vasculitis crioglobulinemia asociada a la hepatitis C la administración del interferón alfa está indicado y la virabidina.
Púrpura de Henoch – Schonlein
La púrpura de Henoch . Schonlein es la vasculitis sistémica más común en los niños. Se caracteriza por el depósito de complejos inmunes con el dominio de la IgA en las paredes de los rasos de arteriolas, vénulas y capilares tiene un pico a los cinco años y la enfermedad se produce después de un episodio de una infección respiratoria alta. La púrpura palpable, las artralgias y el dolor abdominal son las manifestaciones más importantes. La mitad de los pacientes puede tener hematuria y proteinuria, pero solo el 10 al 20% pulmonar y las neuropatías pueden presentarse.
La púrpura de Henoch – Schonlein:
Se puede presentar en adultos jóvenes y en la tercera edad tanto en hombres como en mujeres, generalmente existe un antígeno precipitante como una inflamación, picadura de insectos, medicamentos, tanto en los niños como en el adulto el tratamiento a base de corticoides e inmunosupresores produce controversias pero existen dos problemas básicos tanto en niños como en adultos y son el compromiso abdominal (posibilidad de hemorragia gastro – intestinal, perforación, perforación de asas intestinal y obstrucción en niños, como también el compromiso renal en estas situaciones el uso de corticosteroides y azatiopirina, parece que es beneficioso para los pacientes. Ya que si no se tratan algunos pacientes con glomerulonefritis, la evolución a una nefropatía rápidamente progresiva se puede producir; otra indicación para los inmunosupresores es cuando encontramos compromiso pulmonar y/o neuropatía.
El abuso que se ha tenido con la denominación vasculitis de pequeños vasos asociado a ANCA que se ha observado en un grupo de pacientes que consultaron por dolor abdominal, púrpura palpable y nefritis no necesariamente se trata de una púrpura de Henoch – Schonlein, estos pacientes pueden tener una poliangeitis microscópica, una granulomatosis de Wagener, un síndrome de Churg – Strauss y/o una vasculitis inducida por medicamentos asociada a ANCA y requiere tratamiento inmunosupresor y son de pronóstico reservado si no se realiza un diagnóstico oportuno y por ende con tratamiento adecuado.
En algunos pacientes con problemas renales se pueden presentar casos refractarios al tratamiento.
Hepatitis C y crioinmunoglobulinemia
El virus de la hepatitis C es un virus RNA de simple cadera, que puede causar una infección aguda y crónica. La prevalencia varía de acuerdo a la región geográfica. En USA aproximadamente 17.500 personas adquieren el virus anualmente. La mayoría de los cuales desarrollan la infección crónica. El origen de la infección es indeterminada en el 40 a 50% de los casos. El HCV se transmite parenteralmente, con una alta tasa de infección entre la población de alto riesgo como los drogadictos que utilizan drogas IV, y los hemofílicos. El riesgo de infección en los últimos años a través de la transfusión sanguínea se ha reducido.
En Colombia y en la mayoría de los países de América Latina un control estricto de los bancos de sangre empezó a realizar después de 1993, por ellos muchos pacientes quedaron expuestos a este virus, por lo que el cuadro clínico se empieza a presentar en estos años venideros. El virus además de ser linfotrópico causando una intensa estimulación del sistema inmunitario, por ello la respuesta inmunitaria es diversa y variada. Entre al respuesta humoral se describe el factor reumatoideo en el 76% de los casos, anticuerpos antimúsculo liso en el 66% de los casos, anticuerpos antinucleares ocurre entre el 14 al 22% y crioglobulinas se puede detectar en el 36% de los casos. Anticuerpos antimitocondriales, anti hígado/riñón son raros, pero sin embargo se puede encontrar en el 41% de los casos anticuerpos anti – tisular.
La principal manifestación extra-hepática es la crioglobulinemia mixta. Estudios sobre evolución de la crioglobulinemia mixta son escasos. La mortalidad se encuentra entre el 44% y el 62% de las diferentes series. La causa más frecuente de muerte es la insuficiencia renal, la falla hepática, la enfermedad cardiovascular y las enfermedades linfoproliferativas.
Considerar muchos de estos pacientes refractarios al tratamiento.