La circulación pulmonar responde a la hipoxia con incremento de la presión en la arteria pulmonar y aumento de las resistencias vasculares, a diferencia de la circulación sistémica la cual responde con vasodilatación.
La respuesta se inicia segundos después de la exposición a la hipoxia, manteniéndose por minutos o hasta horas de acuerdo a la intensidad del estímulo (los niveles necesarios para desencadenar la respuesta oscilan alrededor de una presión alveolar de 60 mm Hg) con cambios fisiológicos como el incremento de la frecuencia respiratoria y de la presión de la arteria pulmonar y con cambios variables en la presión auricular izquierda, la frecuencia cardíaca y el gasto cardíaco.
Los sensores por medio de los cuales se detecta la disminución en el contenido de oxígeno, clásicamente reconocidos, son las células musculares lisas que producen vasoconstricción inmediata, el glomus celular del cuerpo carotídeo liberando dopamina y los cuerpos neuroepiteliales que secretan en forma rápida y organizada serotonina que va a ser la directa implicada en la generación de corrientes iónicas a través de las membranas celulares favoreciendo el influjo de Calcio y la salida de Potasio.
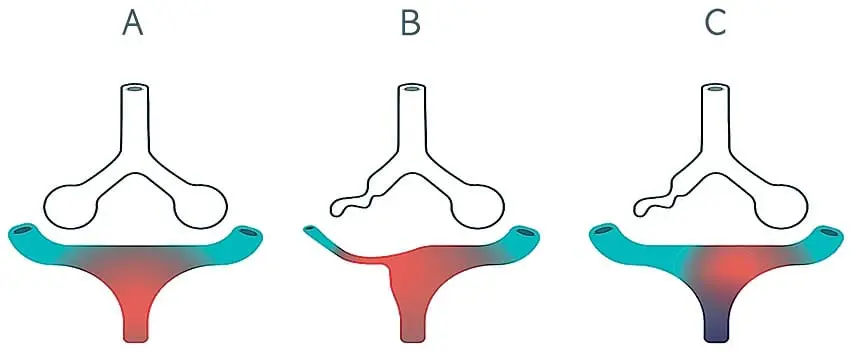
La vasoconstricción pulmonar hipóxica es un método de adaptación para redistribuir el flujo de áreas pobremente ventiladas a zonas donde exista una mejor relación V/Q, minimizando la hipoxemia. Generalmente estas zonas corresponden a los ápices pulmonares con el reclutamiento de capilares alveolares, considerando la menor perfusión con relación a las bases, consecuencia de la menor presión pulmonar por el efecto de la gravedad.
Fabio Barón, MD*,
Oscar A. Sáenz, MD**
* Residente III Medicina Interna. Neumología
** Neumológo Director Dpto Fisiología Pulmonar Hospital Santa Clara
Señales intracelulares en la vasocostricción pulmonar hipóica
La Protein kinasa C, una enzima intracelular, Ca dependiente, al ser activada estimula la síntesis de DNA en la capa muscular de las arteriolas pulmonares (sitio anatómico donde ocurre la VPH) e influencia el intercambio de Na, k, y Ca. La PKC es activada por el diacilglicerol, el cual es generado por la hidrólisis del fosfatidil inositol 4,5 bifosfato (PIP2).
El otro producto de esta reacción, el inositol 1,4,5 trifosfato (IP3) se une al receptor en el retículo endoplásmico, permitiendo la liberación del Ca intracelular y activando los mecanismos que llevarán al desarrollo de la VPH. La hipoxia promueve la liberación y proliferación del IP3 en los fibroblastos de las arterias pulmonares. (Gráfica 1) (Lea también: Síndrome de lesión pulmonar asociada a la ventilación mecánica)

Gráfica No. 1
Mediadores en la vasoconstricción pulmonar hipóica
Muchos mediadores han sido involucrados en el mecanismo de respuesta de la VPH, sin embargo ninguno de ellos explica en forma convincente los eventos fisiopatológicos que generan esta respuesta, sugiriendo que son múltiples los mecanismos y mediadores involucrados.
Además llama la atención, cómo en los extremos de la hipoxia, la VPH es ineficiente y los mismos mediadores pueden tener respuestas variables, como por ejemplo la norepinefrina, que posee un tono bifásico, actúa como vasoconstrictor si el tono de la circulación pulmonar es bajo o como vasodilatador si la respuesta vasoconstrictora y el tono son elevados.
( Gráfica 2)

Gráfica No. 1
Mitogénesis e hipoxia
La hipoxia incrementa la proliferación de los tres tipos celulares de la pared arterial; muchos de los factores involucrados en la respuesta vasoconstrictora como la endotelina, el factor de crecimiento derivado de las plaquetas, el factor de crecimiento vascular endotelial y la serotonina, tienen además la capacidad de estimular el crecimiento de las células musculares.
Relevancia clínica de la vasoconstricción pulmonar hipóica VPH
Las implicaciones clínicas no han sido claramente entendidas, pero en general se aceptan tres tipos de respuesta:
Pérdida de la respuesta en estados de lesión pulmonar
Patologías como el SDRA en el cual la hipertensión pulmonar encontrada se debe a una serie de factores vasculares como la obstrucción, la obliteración y la misma vasoconstricción secundaria a la liberación de endotelinas, prostaglandinas y radicales libres, perpetúan una respuesta en la que los capilares en su mayoría no pueden ser reclutados, aumentando el grado de hipoxemia, lo que en últimas lleva la curva a uno de sus extremos con la consecuente disminución o pérdida total de un mecanismo compensatorio como la VPH.
Disminución o atenuación de la vph despues la exposición crónica a la hipoxia:
La hipoxia crónica se asocia a cambios morfológicos en la vascularización pulmonar con aumento especialmente de la capa muscular en las pequeñas arteriolas (ver mitogénesis e hipoxia) lo que conlleva una elevación persistente de la presión de la arteria pulmonar y de la resistencia vascular.
Estas alteraciones también desencadenan variaciones en los potenciales de membrana, aumentando en forma marcada su umbral de respuesta a la hipoxia y disminuyendo el influjo iónico especialmente de Calcio. El mecanismo por el cual se generan estas variaciones permanece desconocido.
Alteración de la respuesta en otros estados clínicos:
Medicamentos: Diversos fármacos antagonizan el efecto de los mediadores involucrados en el origen de la VPH (Anestésicos inhalados, Nitroprusiato, Calcio antagonistas, Inhibidores de la COX, Beta bloqueadores no cardioselectivos, Xantinas.)
Atelectasias-Neumonías: Se encuentra un marcado aumento en la producción de sustancias vasodilatadoras como las prostaglandinas en el área afectada impidiendo los mecanismos compensatorios de redistribución del flujo.
EPOC-TEP recurrente: Al parecer la hipoxia crónica genera los mecanismos ya comentados impidiendo la VPH en forma parcial.
Cirrosis hepática: En estados terminales se encuentra completamente abolida la respuesta a la hipoxia, pero el mecanismo por el cual se impide la respuesta no está aclarado, aunque se involucran sustancias como las prostaglandinas.
Conclusión
La VPH es un mecanismo compensatorio que favorece la disminución del cortocircuito generando el reclutamiento de nuevos capilares especialmente apicales y favoreciendo así la disminución de la hipoxemia. Sin embargo esta explicación es sólo parcial pues los mecanismos y mediadores involucrados no se han aclarado totalmente.
Bibliografia
1. Jones, R and L. Reid. 1995. Vascular remodelling in clinical and experimental pulmonary hypertensions. Pulmonary Vascular Remodelling. Portlandpress, London. 47-115.
2. Staub. N. C. 1985. Site of hypoxic pulmonary vasoconstriction. Chest 88:240S-245S.
3. Fishman, A. P. 1961. Respiratory gases in the control of the pulmonary circulation. Physiol. Rev. 41:214.
4. Fishman, A. P. Pulmonary Circulation. In Handbook of Physiology: The Respiratory Sistem I. American Physiological Society. Bethesda. MD. 93-165.
5. Furchgott. R. F. 1985. Interactions of endothelial cells and smooth muscle cells of arteries. Chest 88:210S-213S.
6. Weir, E. K, 1985. Pulmonary vascular reactivity. Chest 88(Supp. 4): 199S-272S.
7. Gossage, J: R. and B. W. Christman. 1994. Mediators of acute and chronic pulmonary hypertension.. Seminars in Respiratory Medicine 15:190-198, 452-453.
8. Ryan, U. S. 1990. Receptors on pulmonary endothelial cells. Am. Rev. Respir. Dis. 141:S132-S136.
9. Rubin, L. J. 1997. Primary pulmonary hypertension. N. Engl. J. Med. 334: 329-336.
10. D´Alonzo, G. G. R. J: Barst, S. M. Ayres et al, 1989. Survival in patients with primary pulmonary hypertension: results from a national retrospective registry. Ann. Intern. Med. 115:343-349.
11. Baugh CW, Cornatt RW, Hatcher JD. The adrenal gland and the cardiovascular changes in acute anoxic anoxia in dogs. Circ Res 1959;7:513-520.
12. Gorlin R, Lewis BM. Circulatory adjustments to hypoxia in dogs. J Appl Phsiol 1954;7:180-185.
13. Harrison TR, Blalock A. The regulation of the circulation. VI. The effects of severe anoxaemia of short duration on the cardiac output of morphinised and trained unnarcotized dogs. Am J Physiol 1927;80:169-178.
14. Sidi D, Kuipers JRG, Teitel D, et al. Developmental changes in oxygenation and circulatory responses to hypoxaemia in lambs. Am J Physiol 1983;245:H674-H682
15. Weir EK, Archer SL. The mechanism of acute pulmonary vasoconstriction: the tale of two channels. FASEB 1995;9:183-189.
16. Von Euler US, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand 1946;12:301-320.
17. McMurtry I, Davidson A, Reeves J, et al. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ Res 1976;38:99-104.
18. Maden J, Dawson C, Harder D. Hypoxia-induced activation in small isolated pulmonary arteries from the cat. J Appl Physiol 1985;59:113-118.
19. Madden JA, Vadula MS, Kurup VP. Effects of hypoxia and other vasoactive agents on pulmonary and cerebral artery smooth muscle cells. Lung Cell Mol Physiol 1992;7:L384-L393.
20. Vadula MS, Kleinman JG, Madden JA. Effects of hypoxia and norepinephrine on cytoplasmic free Ca2+ in pulmonary and cerebral arterial myocytes. Am J Physiol 1993;265:L591-L597.
21. Bennie RE, Packer CS, Powell DR, et al. Biphasic contractile response of pulmonary artery to hypoxia. Am J Physiol 1991;263:L156-L163.
22. Lloyd TC. Pulmonary vasoconstriction during histotoxic hypoxia. J Appl Physiol 1965;20:488-490.
23. Fidone S, Gonzalez C, Yoshizaki K. Effects of low oxygen on the release of dopamine from the rabbit carotid body in vitro. J Physiol 1982;333:93-110.
24. Ganfornina MD, Lopez-Barneo J. Single K+ channels in membrane patches of arterial chemoreceptor cells are modulated by O2 tension. Proc Natl Acad Sci USA 1991;88:2927-2930.
25. Post J, Hume J, Archer SL, et al. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol 1992;262:H882-H890.
.