Discusión
La Proteinosis Alveolar (PA), corresponde a una entidad rara, en la cual los alvéolos y unidades respiratorias se llenan progresivamente de un material lipoproteináceo, que con el tiempo, si no es retirado de los pulmones, podrá llevar al paciente a insuficiencia respiratoria. Fue descrita por primera vez por Rosen en junio de 1958 (5), reportando 24 casos de fosfolipoproteinosis alveolar en los Estados Unidos, y tres casos de Canadá, Inglaterra e Italia uno cada país respectivamente (2).
La entidad se ha descrito en todas las edades tratándose de un desorden siempre fatal en el recién nacido conocido como la forma congénita (1). Suele presentarse más frecuentemente en hombres, en una relación de 3 a 1.
Habiéndose descrito tres variedades de presentación a saber: la más común la idiopática del adulto, de causa probablemente autoinmune, la variedad neonatal y la forma secundaria asociada a componentes exposicionales, neoplasias hematológicas o desórdenes mieloides (3 – 4). (Lea también: Validación de una Escala para Predecir la Mortalidad por Neumonía Adquirida en la Comunidad (CURB-65)
Primer caso de fosfolipoproteinosis alveolar
Corresponde a un paciente de sexo masculino, de ocupación comerciante, de 38 años de edad, quien consulta a la unidad de Neumología del Hospital de San José, en el año de 1994, por un cuadro de más o menos 9 meses de evolución, posterior a episodio gripal severo, caracterizado por tos intermitente de tipo irritativo ( No productiva), acompañado inicialmente de disnea no sibilante intermitente, para lo cual fue manejado por médico particular con antibiótico terapia (Trimetroprim sulfa ) y antihistamínicos con poca mejoría, posterior a tres meses de su inicio la disnea se hace permanente y progresiva hasta clase funcional II – III, razón por la cual acude a nuestra institución.
Niega factores de riesgo claros para enfermedad pulmonar, salvo un tabaquismo de seis paquetes al año hasta cinco meses antes del día de la consulta. Niega antecedentes patológicos o quirúrgicos. No hay otros antecedentes de importancia.
Al examen físico de ingreso se encuentra un paciente en regular estado general, algo enflaquecido, disneico con leve cianosis labial. TA 130/80, FC: 106 FR: 24, Saturación 84%. La auscultación cardiopulmonar se consideró dentro de límites normales, sin agregado aparentes. El resto del examen físico normal.
Estudios de paraclínicos
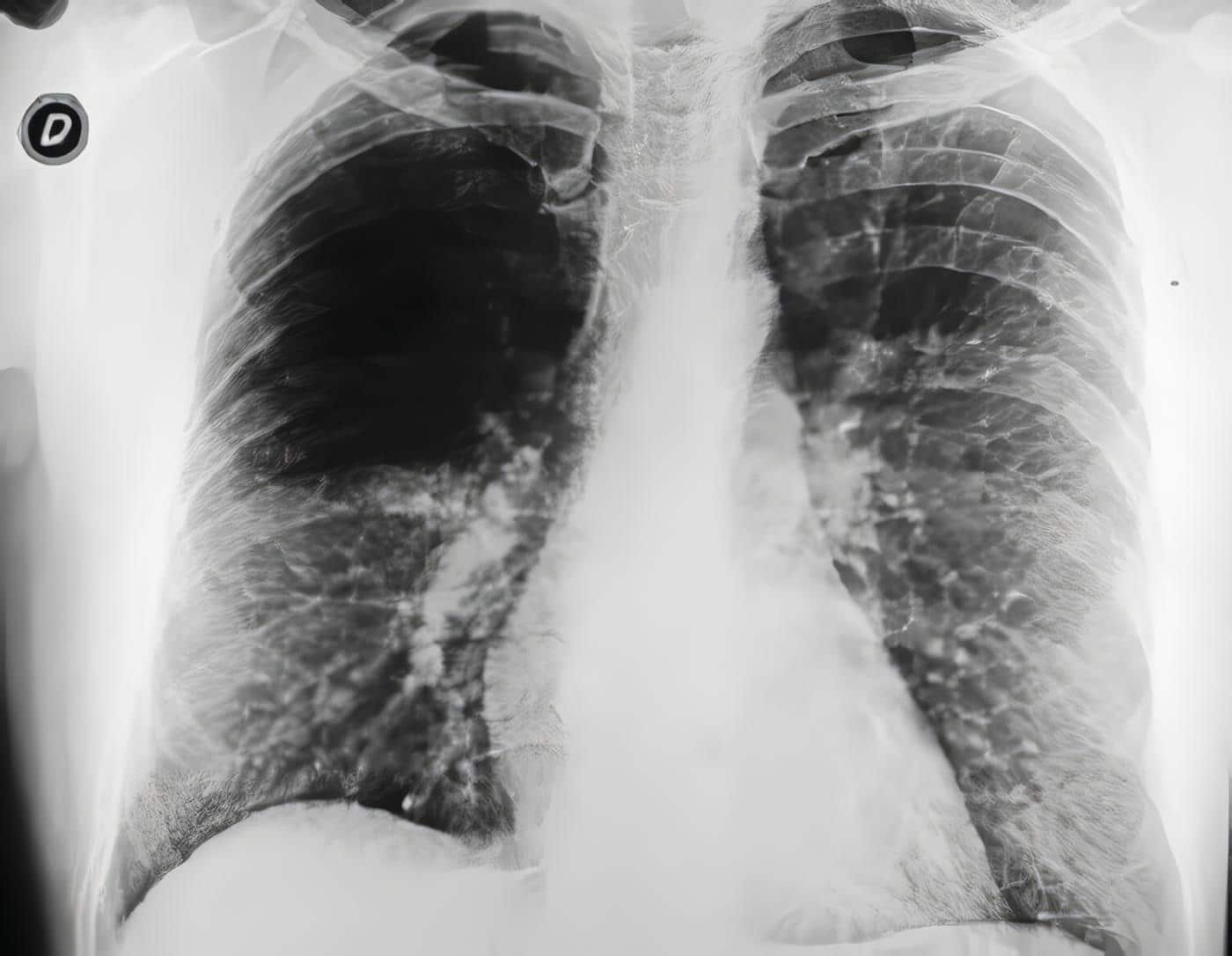
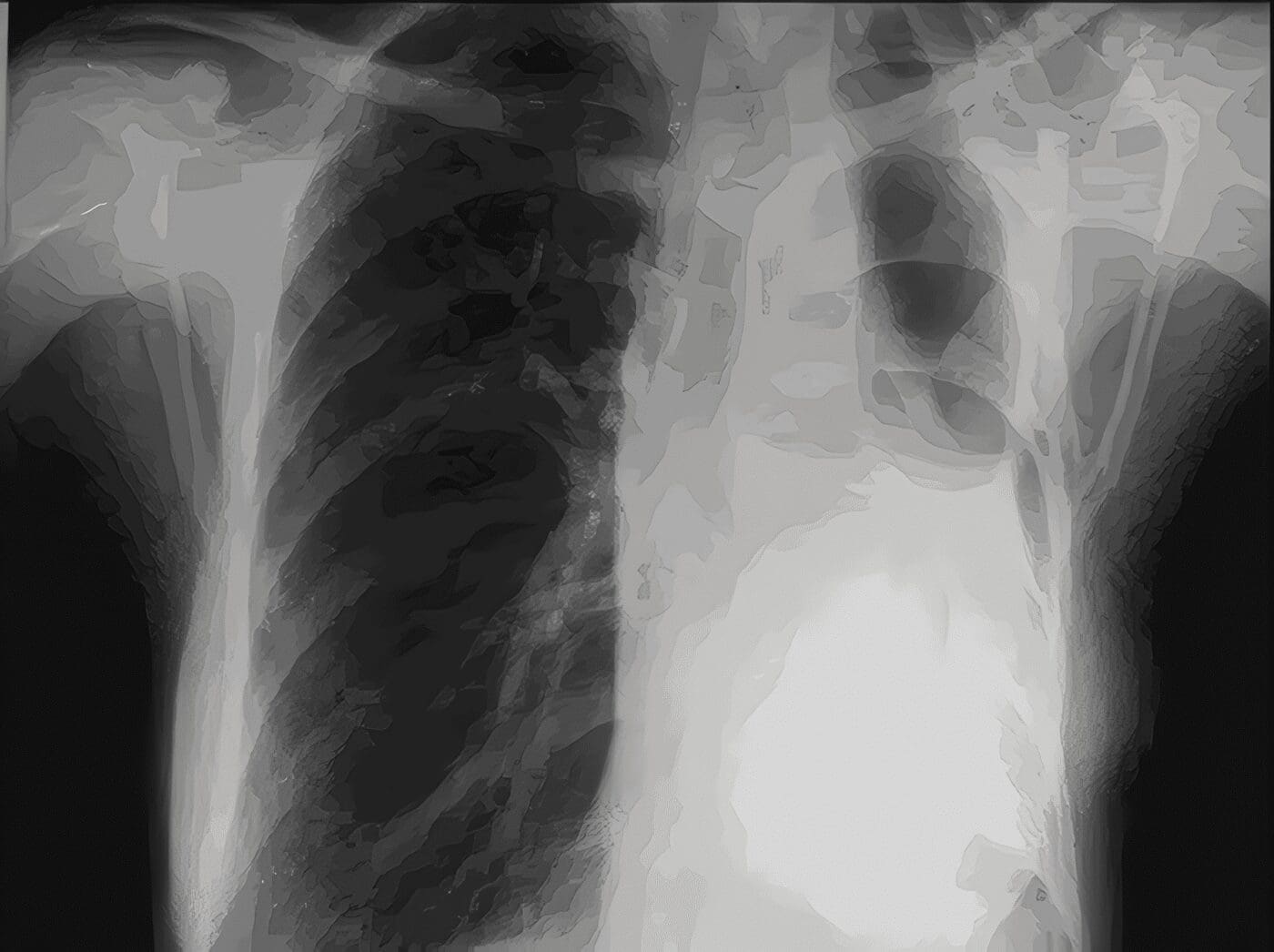
Cuadro hematico: leucocitos 7800 – PMN 51% – Linfocitos 32% – Eosinofilos 3% – Monocitos 4% – Hb 13,4 – VSG : 22 mm, glicemia, BUN, creatinina en rangos normales. Gases arteriales mostraron hipoxemia ( PaO2 : 48 ) , con leve alcalosis respiratoria ( Ph : 7, 44 – Pa CO2 : 26 ) , la placa de tórax ( Figura 1) mostró pobre esfuerzo inspiratorio, con infiltrados de ocupación alveolar parchados difusos bilaterales, de localización principalmente hacia las bases.
Se realizaron muestras de esputo inducido Nº 2 para Bk negativos, tinción de Gram mostró cocos Gram positivos (+) bacilos Gram negativos (-), escasa reacción leucocitaria. Se decide llevar al paciente a fibrobroncoscopia óptica, obteniéndose en el BAL 110 cc de líquido lechoso espeso, del cual las tinciones especiales para gérmenes comunes, hongos, Bk fueron negativas, la tinción PAS fue positiva.

Figura 1. Rx de tórax inicial, mostrando infiltrados alveolares difusos, bilaterales, principalmente en dos tercios inferiores.
Con diagnóstico de proteinosis alveolar el paciente es llevado a lavado pulmonar total (con diferencia de 12 días entre pulmón y pulmón), con controles gasimétricos 10 días post lavados que mostraron PaO2 : 62 Saturación de 93%, PaCO2 : 28, el control radiológico un mes después del lavado mostró resolución significativa de los infiltrados alveolares descritos inicialmente, (Figura 2 ), pruebas espirométricas completamente normales, clínicamente resolución completa de la disnea.

Figura 2. Rx de tórax 14 días post lavado, muestra significativa mejoría, y resolución de los infiltrados.
Segundo caso de fosfolipoproteinosis alveolar
Corresponde a un paciente de 26 años de edad, pastor evangélico que, consulta a nuestra institución en el año de 1999, por un cuadro de 12 días de evolución, caracterizado por malestar general, astenia y adinamia, fiebre no cuantificada, tos con expectoración en cuatro ocasiones hemoptoica, disnea clase funcional II.
Al interrogatorio se establece la presencia de cuadro similar tres meses antes de su consulta actual que fue interpretado por médico particular como bronquitis aguda, para lo cual recibió tratamiento antibiótico que no recuerda. Niega antecedentes de importancia, al igual que niega exposicionales.
Al examen físico de ingreso se encontró un paciente en regular estado general, TA : 130 / 70, FC 118, FR 28, Saturación 80%. Temperatura 37,8°.
Los ruidos cardíacos taquicárdicos sin agregados, los ruidos respiratorios disminuidos en forma generalizada, con escasos estertores inspiratorios bilaterales, esporádicas sibilancias de final de espiración, el abdomen blando depresible normal, las extremidades mostraban cianosis ungueal.
Exámenes médicos
El cuadro hemático mostró leucocitosis de 11.150, 58%, Linfocitos 39%, monocitos 3%, VSG : 14 mm , PCR : 7 – Gases arteriales PaO2 : 42% , PaCO2: 28 mm , HCO3: 17 , Saturación: 80 %, la química sanguínea completamente normal.
Los Rx de tórax mostraron infiltrados alveolares difusos bilaterales, con áreas tendiente a la confluencia.
Se consideró inicialmente una neumonía grave bilateral. Por consiguiente se inició manejo con base en Claritromicina más Ceftriaxone y medidas de soporte. Cuatro días posterior al inicio de la terapia el paciente continúa comprometido clínicamente, hemocultivos negativos. Se decide entonces realizar Fibrobroncoscopia la cual fue normal, salvo la presencia de BAL moderadamente hemorrágico, los reportes de los lavados y cepillados para gérmenes comunes, hongos y BK fueron informados negativos.
Los estudios de patología solamente mostraron un BAL hemorrágico con diferencial de 70% macrófagos alveolares, 25 % mononucleares, 5% eosinófilos.
Se consideró con este reporte la posibilidad de neumonía eosinofílica vs. hemorragia alveolar. Asimismo, se solicitaron estudios de inmunología, y se decide llevar al paciente a biopsia abierta de pulmón. Esta mostró alvéolos llenos de material proteinaceo PAS positivo, altamente sugestiva de Fosfolipoproteinosis Alveolar ( Figura 3 )
Diagnóstico con el cual el paciente se llevó a lavado pulmonar total en dos ciclos con diferencia de 12 días entre uno y otro pulmón. Procedimiento durante el cual se obtuvo posterior a 15 lts de SSN un material con las características propias de la entidad. (Figura 4).
El paciente posterior a los procedimientos recupera su función pulmonar, con controles posteriores durante cuatro años con estabilidad clínico – radiológica y funcional.

Figura 3. Obsérvese los alvéolos inundados de un material PAS Positivo, que corresponde a lipoproteinas acumuladas.

Figura 4. Material decantado, obtenido en el lavado pulmonar total de uno de nuestros pacientes.
Tercer caso de fosfolipoproteinosis alveolar
Corresponde a un hombre de 34 años de edad, de ocupación limpiador de chimeneas y calderas por los últimos 10 años, quien consulta a nuestra institución en enero de 1997, refiriendo un cuadro de más o menos 15 meses de evolución caracterizado por tos irritativa de predominio vesperal , y disnea progresiva hasta Clase Funcional II , episodios ocasionales de disnea sibilante para lo cual había recibido anteriormente inhaloterapia.
Además de su factor de riesgo exposicional laboral refería tabaquismo de siete paquetes año hasta ocho meses antes de la consulta, no otros antecedentes de importancia.
El examen físico un paciente en aceptable estado general: TA: 100/80, FC: 80, FR: 21, Sat: 87%.
Presencia de cianosis labial leve, los ruidos cardíacos rítmicos sin agregados. Los ruidos respiratorios disminuidos en forma generalizada sin agregados por anotar. El abdomen normal, y las extremidades con cianosis ungueal leve.
Exámenes clínicos
El cuadro hemático y la química sanguínea completamente normales. Los gases arteriales mostraban PaO2 : 51 – PaCO2 : 33 mm – HCO3: 19 – Sat: 85%, la placa de tórax mostraba un infiltrado alveolar que comprometía los dos tercios inferiores de ambos campos pulmonares, con imágenes de TAC de Tórax que mostraban característico infiltrado alveolar y en vidrio esmerilado parchado difuso bilateral (Figura 5 ).
La curva Flujo Volumen mostró una alteración ventilatoria restrictiva moderada comprometiendo levemente los volúmenes y flujos pulmonares, con respuesta limítrofe del VEF1 al broncodilatador.
El paciente se llevó a Fibrobroncoscopia óptica, procedimiento durante el cual se obtuvo un BAL típicamente lechoso con Bx transbronquial. Esto confirmó el material proteináceo inundando las unidades respiratorias, característico de la PAP.
El paciente se llevó a Lavado Pulmonar Total. Procedimiento que se tuvo que realizar en cuatro ocasiones con diferencia de 7 meses cada uno por evolución rápida de las manifestaciones clínico funcionales, hasta obtenerse estabilidad del cuadro por año y medio hasta el momento. Llamativamente el paciente estabiliza su cuadro cuando decide retirarse en forma definitiva de su exposición laboral.

Figura 5. TAC de tórax de alta resolución. Obsérvese el infiltrado parchado en vidrio esmerilado comprometiendo ambos campos pulmpnares.
Patogénesis
Se han propuesto dentro de la fisiopatología de la entidad tres eventos que pueden explicar la razón por la cual dicho material se acumula a nivel alveolar , la forma neonatal que obedece principalmente a un defecto en la producción de proteína B ( SP-B ) del surfactante la cual se codifica en el cromosoma 2, deficiencia que lleva a un mal-funcionamiento en la tensión superficial alveolar y un favorecimiento en el depósito de estos fosfolípidos en las unidades respiratorias del paciente pediátrico, llevando al mismo a una muerte en un período no mayor a seis meses (1- 3).
Esta situación se transmite de forma autosómica recesiva, y en individuos homozigotos es fatal, mientras que en aquellos heterocigotos puede ser asintomática (1).
El otro evento referido en la literatura médica, y de reciente conocimiento, obedece a una deficiencia en la respuesta del factor estimulador de colonias de granulocitos y macrófagos (GM-CSF), como resultado de la presencia de anticuerpos circulantes contra dicho factor, o una disrupción en la superficie celular de los receptores para el GM-CSF (1, 4, 6).
Histopatología
Característicamente los estudios histopatológicos, revelan la presencia de alvéolos llenos de un material proteináceo PAS positivo, hiperplasia del epitelio alveolar, en algunos pacientes se describe engrosamiento de los septos alveolares, y se ha descrito además en algunos individuos focos localizados de neumonitis inespecífica, y en estadios de cronicidad se ha observado claras áreas de fibrosis pulmonar.
Cuando se utilizan tinciones específicas de inmuno-histoquímica, especialmente en las formas congénitas de la enfermedad, se puede detectar la ausencia de la proteína B del surfactante (SP-B), por el contrario se detecta grandes cantidades de depósito de las proteínas A y C (SP-A, SP-C) ( 1
Diagnóstico
El diagnóstico de la entidad, requiere de un enfoque clínico, radiológico, funcional e invasivo (broncoscópico – biopsia abierta).
Desde el punto de vista clínico, la presentación de la entidad suele ser crónica, siendo el síntoma principal la disnea, que se presenta en más del 90% de los casos, disnea que suele ser progresiva y en la medida en que se retarde el diagnóstico por consiguiente la terapia, el resultado final será en la gran mayoría de los casos la insuficiencia respiratoria, y en una minoría la fibrosis pulmonar.
Las imágenes son de mucha utilidad principalmente cuando hablamos de la TAC de alta resolución, la cual nos muestra infiltrados en vidrio esmerilado y alveolares parchados difusos bilaterales, que característicamente se suelen localizar en las zonas dependientes del pulmón esto es, campos medios e inferiores y segmentos posteriores a nivel superior.
Las pruebas funcionales mostraran los resultados esperados en una patología que afecta de la forma descrita las unidades respiratorias , mostrando a nivel gasimétrico hipoxemia de leve moderada en la mayoría de los casos con compromiso de la diferencia alvéolo arterial.
Por su parte, las pruebas espirométricas mostrarán alteración ventilatoria restrictiva de leve a severa según sea el caso, en muchas situaciones comprometiendo secundariamente los volúmenes y flujos. Las pruebas de difusión (DLCO) mostrarán una disminución significativa de la misma.
La fibrobroncoscopia óptica se convierte en una excelente alternativa diagnóstica ya que en un porcentaje muy alto de los casos el BAL nos permitirá recuperar un material de consistencia espesa y de aspecto lechoso característico del material que inunda los alvéolos. La biopsia transbronquial y rara vez necesaria la biopsia abierta pulmonar identificará los alvéolos llenos por el material ya descrito (ver descripción histopatológica).
Tratamiento
El propósito de la terapia radica en la posibilidad de retirar el material proteinaceo que llena los alvéolos, para lo cual la técnica más efectiva es el lavado pulmonar total, procedimiento descrito desde el año 1963, por Ramírez Raya Rivera.
Procedimiento que se realiza bajo anestesia general y requiere una experiencia por parte del anestesiólogo en la intubación orotraqueal con tubo de doble lumen, intubación confirmada broncoscópicamente, permitiendo por una parte ventilar al paciente por un pulmón e ir lavando el pulmón contralateral, utilizando solución salina normal a temperatura corporal, para lo cual se instila de 10 a 20 o incluso hasta más litros de solución durante el procedimiento (5).
Pasados 10 a 15 días se procederá a lavar el otro pulmón. Con este lavado gran parte del material es retirado, y algunos consideran que en algunos individuos puede retirarse la noxa generadora de la entidad.
Otra alternativa terapéutica es la utilización de terapia con factor activador de colonias de granulocitos y monocitos (GM-CSF), terapia que se hace efectiva especialmente en aquellos pacientes en quienes se logra detectar anticuerpos contra el GM-CSF, algunos expertos considera que debiera asociarse a plasmaféresis para disminuir los títulos de anticuerpos (6).
Algunas publicaciones que probablemente no dejan de ser más que anecdóticas, hablan sobre la utilización crónica (por más de seis meses) de dosis diarias de 45 Mg. de GM-CSF, en pacientes que por su edad o patologías asociadas no toleran un procedimiento de lavado pulmonar , y aparentemente con resultados muy satisfactorios (8).
No obstante en opinión del autor en este grupo de pacientes con contraindicaciones relativas para un lavado pulmonar, podría considerarse la utilización de lavados lobares con la utilización broncoscópica, como lo manifiestan algunas publicaciones de la literatura médica mundial (9).
Bibliografia
1. Daphne E. De Mello. Pulmonary Alveolar Proteinosis : A Review. Pediatric Pathology and Molecular Medicine 2001;20: 413 – 432.
2. James L. Knott, et al. Pulmonary Alveolar Proteinosis. Annals of Internal Medicine 2001; Sept 1961; Vol 55 , N 3 : 481- 490.
3. Mani S. Kavuru .- Marc popovich. Therapeutic Whole Lung Lavage : A Stop Gap therapy for Alveolar Proteinosis. Chest Oct, 2002; Vol 122 N 4: 1- 2 .
4. Yoshioca Y. Increased Circulating CD 16 + CD 14dim monocytes in a patient with Pulmonary Alveolar Proteinosis. Respirology – Sept 2002; 01: 273-279.
5. Jean S. Bussiéres. Whole Lung Lavage. Anesthesiology Clinics Of North America, Sept 2001; Vol 19 , N 3 : 1- 10.
6. Bonfield TL , et al. Anti GM-CSF Titer Predicts response to GM-CSF Therapy in Pulmonary Alveolar Proteinosis. Clinical Inmunology , Dec 2002; 105 ( 3 ): 342- 350.
7. Arbiser ZK. Guidot DM, et al. Pulmonary Alveolar Proteinosis mimiking Idiopathic Pulmonary Fibrosis. Annals Of Diagnostic Pathology, Apr 2003; 7(2):82-86.
8. Hashizume T. Pulmonary Alveolar Proteinosis Successfully treated with Ambroxol. Internal Medicine, Dec 2002, 41 (12): 1175-1178.
9. Shih-Lung Cheng. et al. Pulmonary Alveolar Proteinosis: Treatment By Bronchofiberscopic Lobar Lavage.Chest, Oct 2002; 122(4):1480-1484.