Guía de Estudio y Manejo
Carlos J. Montoya, Helí Salgado, Julieta Henao,
Julio C. Orrego, Pablo J. Patiño.
Grupo de Inmunodeficiencias Primarias,
Facultad de Medicina – Universidad de Antioquia.
I. Criterios clínicos de sospecha
- Cuadros de infección recurrente anormal causada por gérmenes catalasa positivos y por algunos oportunistas que afectan la piel, mucosas y tejidos profundos. Característicamente, las lesiones presentan alteraciones en la respuesta inflamatoria (supuración anormalmente elevada o ausente). En general, en estos pacientes las infecciones tienen un comportamiento francamente anormal al evaluar su respuesta al tratamiento, diseminación y complicaciones.
- Edad de inicio: En la mayoría de los casos severos, las infecciones se inician durante el primer año de vida. En las formas autosómicas recesivas de la enfermedad granulomatosa crónica, se pueden presentar casos de inicio tardío y menor severidad clínica.
- Otros signos clínicos que deben despertar una alerta inmediata dirigida a descartar una deficiencia en las células fagocíticas son: linfadenopatía crónica y/o supurativa; retardo o mala cicatrización de las heridas; úlceras necróticas no supurativas; caída tardía del cordón umbilical (mayor de 20 días); discromías (albinismo oculocutáneo parcial) y fotofobia; gingivoestomatitis severa y periodontitis; paroniquia recurrente; eczema crónico; abscesos “fríos”; enfermedad inflamatoria intestinal; obstrucción granulomatosa del intestino o aparato urinario; infecciones severas como absceso piógeno hepático o pulmonar, osteomielitis multifocal y/o de huesos cortos; sepsis; neumonías supurativas y neumatoceles perennes.
II. Primera etapa: Estudios básicos de laboratorio
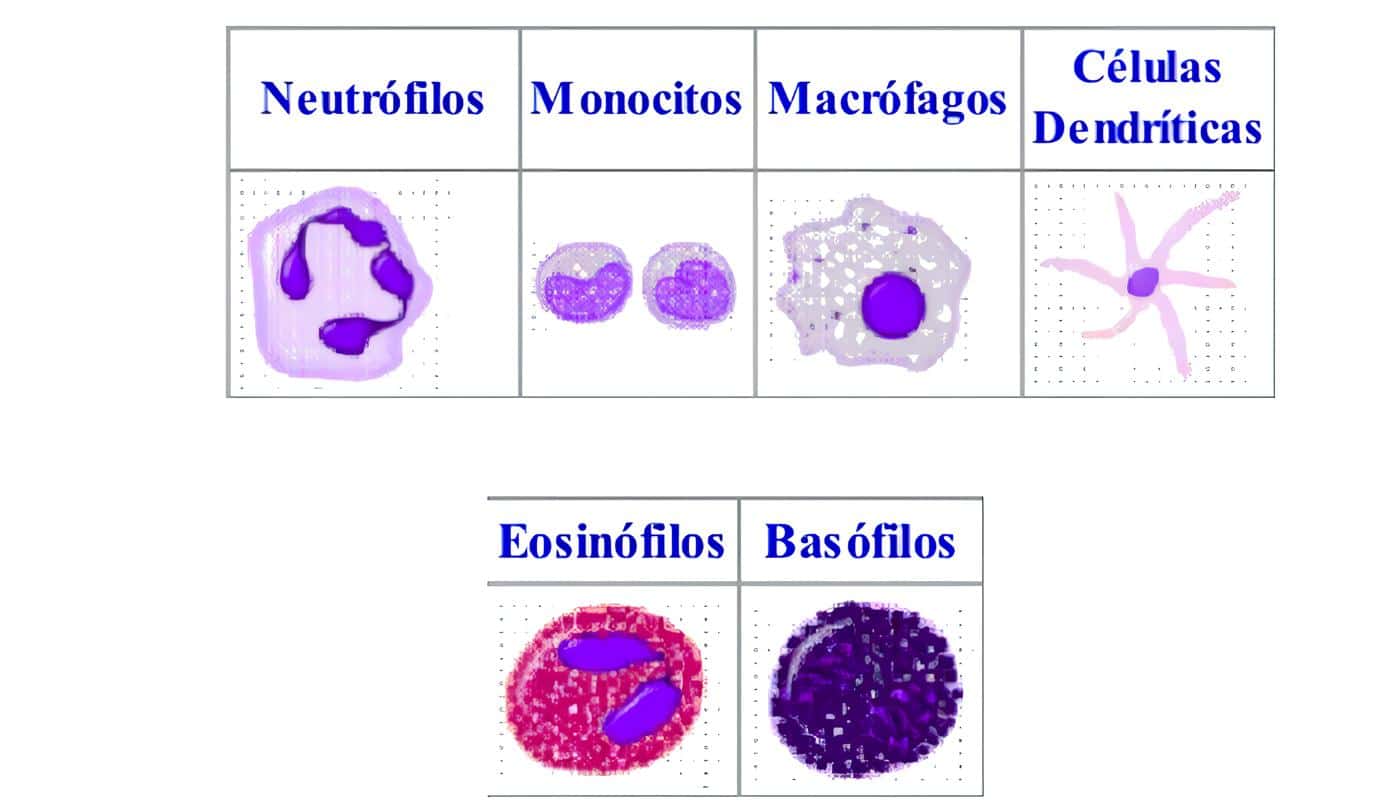
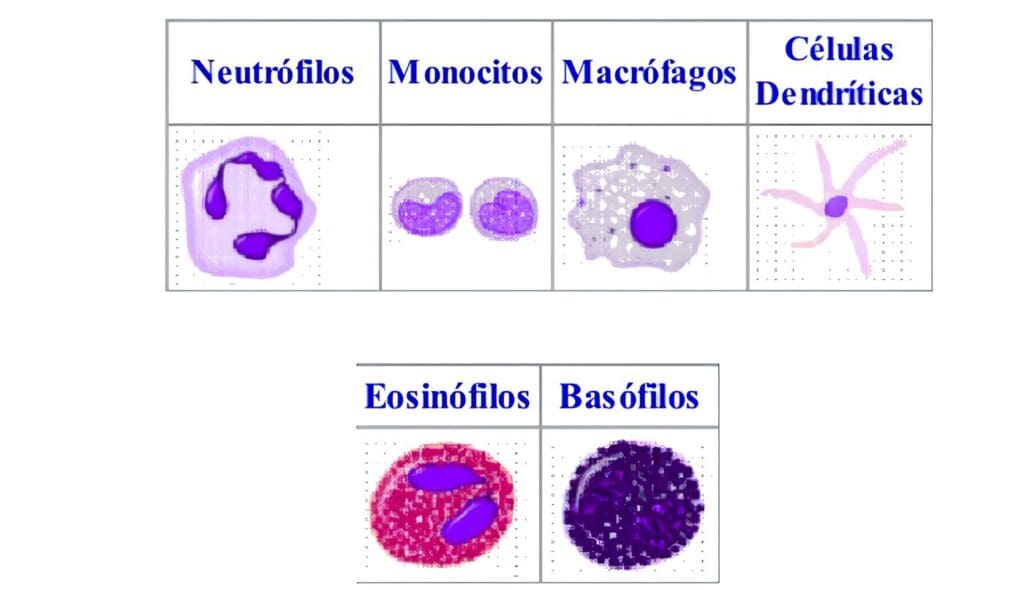
- Hemoleucograma con sedimentación, extendido de sangre periférica y recuento plaquetario. Se debe prestar atención ante la presencia de los siguientes hallazgos, que sugieren el diagnóstico de una deficiencia en las células fagocíticas: granulocitopenia, neutropenia, leucocitosis aún en ausencia de infección, eosinofilia, gránulos anormales en las células de las diferentes series (pero en especial en los granulocitos), anemia moderada a severa, especialmente con características de cronicidad.
- Estudio de la etiología de las infecciones y su sensibilidad. El déficit en la función de las células fagocíticas se asocia con infecciones producidas por: Staphylococcus aureus, Pseudomonas spp, Serratia marsescens, Klebsiella spp, E. coli, Burkholderia cepacia, Proteus spp, Aspergillus spp, Candida albicans, Nocardia spp.En las alteraciones combinadas de la inmunidad celular y los fagocitos, se encuentran infecciones por: Staphylococcus aureus, Haemophilus influenzae, Streptococcus pneumoniae, Candida albicans, Aspergillus spp y bacilos gram negativos.
- Electroforesis de proteínas séricas. Con la determinación de las proteínas totales, su distribución y la relación albúmina/globulinas se establece un importante parámetro del estado nutricional del paciente, para descartar deficiencias de origen carencial. Es común encontrar una hipergamaglobulinemia reactiva ante los procesos infecciosos.
- Estudios imagenológicos cuando sean necesarios (Rx de tórax, TAC, etc.). Se pueden revelar hallazgos como la presencia de neumonías supurativas con desarrollo de neumatoceles y empiemas, abscesos hepáticos.
- Si se confirma un diagnóstico de inmunodeficiencia primaria, se debe realizar el árbol genealógico familiar con el mayor número de generaciones posibles.
III. Estudios de la segunda etapa
- Reducción en placa del Nitroazul de Tetrazolio (NBT) por los neutrófilos de sangre periférica (en reposo y activados con PMA). Evalúa la activación del complejo enzimático NADPH-oxidasa de los fagocitos.
- Si se sospecha una neutropenia cíclica, realizar al menos dos leucogramas por semana, durante 6 semanas.
- Anticuerpos antineutrófilos (anti NA1, NA2, NB1) por inmunofluorescencia indirecta o aglutinación; importantes para descartar una neutropenia autoinmune.
- Biopsia de médula ósea: útil en el diagnóstico de la Disgenesia Reticular y la Enfermedad de Kostmann.
- Cuando se sospecha un Síndrome de Hiper-IgE: se debe realizar dosificación de IgE sérica; en los mayores de 1 año están indicadas las pruebas de hipersensibilidad retardada, por ejemplo 0,1 ml de Candidina intradérmicos; se considera positiva una induración mayor de 5 mm leída a las 48 horas. Debido a que en esta inmunodeficiencia se ha reportado una deficiencia cualitativa en la producción de anticuerpos, se sugiere evaluar también funcionalmente la producción de anticuerpos específicos en respuesta a los antígenos suministrados en la vacunación: para determinar la respuesta ante antígenos proteicos se determinan anticuerpos contra hepatitis B, rubéola o sarampión; para analizar la respuesta a polisacáridos se analizan los anticuerpos contra diferentes serotipos del neumococo. Debido a las implicaciones que tiene una inmunodeficiencia primaria, todo diagnóstico de inmunodeficiencia por alteración en las células fagocíticas debe estar respaldado por una segunda prueba de laboratorio confirmatoria.
IV. Estudios de la tercera etapa
- En los pacientes con sospecha de Síndrome de Hiper-IgE: evaluación funcional por cultivos de células mononucleares de sangre periférica estimuladas con mitógenos y antígenos, para determinar la producción de citoquinas y la proliferación. También se puede realizar cultivo mixto de linfocitos.
- Evaluación de la quimiotaxis in vitro (en gel de agarosa) de los neutrófilos de sangre periférica.
- Estudio de la explosión respiratoria de los neutrófilos: cuantificación de la producción de anión superóxido por espectrofotometría o quimioluminiscencia; determinación de la producción de peróxido de hidrógeno por citometría de flujo (Dihidrorodamina 123).
- Determinaciones enzimáticas: G6PDH, mieloperoxidasa.
- Estudio por citometría de flujo de la expresión de: moléculas de adhesión como las Beta-2 integrinas leucocitarias (CD11/CD18) y la L-selectina, FcR gamma II (CD32), receptores para fragmentos del complemento (CD35, CD11b/CD18). Estos estudios son útiles para descartar entidades como las deficiencias de adhesión leucocitaria.
- Western blot: para evaluar las proteínas del sistema NADPH-oxidasa: gp91-phox, p22-phox, p47-phox, p67-phox.
- Estudios genético-moleculares para la caracterización de mutaciones en las diferentes inmunodeficiencias.
- Cuando estén indicados: caracterización del HLA en los afectados y familiares, y el cariotipo.
V. Manejo terapéutico
1. Medidas generales
Los individuos con deficiencias en la función de las células fagocíticas presentan una notable susceptibilidad al desarrollo de infecciones cuya evolución es más compleja que la observada en los individuos normales. Por ello, además de la terapia antimicrobiana y de reemplazo inmunológico, es fundamental inducir en el paciente y su familia la adopción de hábitos personales y sociales que permitan prevenir el desarrollo de nuevas infecciones o el agravamiento de las presentes. Algunas de las medidas más importantes son:
- El afectado debe portar una tarjeta con la identificación clara de su problema, los riesgos y medidas para una situación de urgencia.
- Los pacientes y sus familiares deben ser advertidos sobre evitar cualquier exposición innecesaria a fuentes de infección (visitas a hospitales, contactos domiciliarios con familiares enfermos, etc.).
- No se recomienda ningún régimen nutricional específico, sin embargo, se debe insistir en una dieta balanceada que cubra plenamente las necesidades en proteínas, vitaminas y oligoelementos
- El paciente no debe fumar ni consumir sustancias ilegales (alcohol, etc.); debe evitar a toda costa constituirse en un fumador pasivo en su medio ambiente familiar y social.
- En los pacientes con deficiencias en las células fagocíticas, se necesita una evaluación semestral por odontología para intervenir las posibles afecciones en la cavidad oral y la instauración de medidas profilácticas destinadas a evitar el desarrollo de periodontitis crónica y otras complicaciones.
- Por lo general, no existen contraindicaciones para la vacunación del paciente o sus familiares cercanos. Sin embargo, debe tenerse en cuenta que cuando existe asociación con una deficiencia cualitativa de la inmunidad celular y/o humoral, la vacunación puede no ser efectiva.
2. Tratamiento
-
El tratamiento ideal de la mayoría de las inmunodeficiencias por alteraciones en la función de las células fagocíticas es el trasplante de médula ósea (o en su defecto, la terapia con sangre del cordón umbilical, ricas en células madres pluripotenciales).
-
Otras alternativas en desarrollo son la terapia génica, los factores de crecimiento de colonias (G-CSF, GM-CSF para los pacientes con neutropenia) y otras citoquinas recombinantes (como el IFN-gamma en la Enfermedad Granulomatosa Crónica).
-
Las infecciones deben recibir una antibioticoterapia temprana y enérgica. Se recomienda la profilaxis permanente con trimetropín-sulfa en la Enfermedad Granulomatosa Crónica, el Síndrome de Chediak-Higashi, el Defecto de adhesión leucocitaria tipo I, la Deficiencia de G6PDH, la Enfermedad de Kostmann y el Síndrome de Hiper-IgE; en este último, también puede utilizarse la profilaxis con dicloxacilina a dosis terapéuticas.
-
Ácido ascórbico: en el Síndrome de Hiper-IgE se recomienda suministrar en dosis de 2 a 4 gramos/día en forma permanente. En el Síndrome de Chediak-Higashi se recomiendan 500 mg/día.
-
Las infecciones mucosas, cutáneas y sistémicas por hongos, deben recibir terapia antimicótica oportuna, idealmente según el reporte del antibiograma; en ocasiones, la prevención de la candidiasis oral requiere el uso repetitivo de enjuagues con Nistatina.
-
Las enfermedades con neutropenia severa y en la Enfermedad Granulomatosa Crónica, tienen valor las transfusiones de leucocitos o granulocitos cuando se presentan infecciones severas de difícil control.
-
En la obstrucción intestinal o urinaria por granulomas de la Enfermedad Granulomatosa Crónica, se recomiendan los esteroides sistémicos (en general, cursos cortos en dosis altas, vía oral).
-
La terapia de reemplazo con gamaglobulina humana venosa sólo estará indicada cuando se demuestre una deficiencia en la producción de anticuerpos específicos antipolisacáridos y el paciente presente un cuadro infeccioso severo que amenace su supervivencia o pueda dejar secuelas graves. En los pacientes sin infección activa (moderada o severa), se recomienda una dosis mensual de gamaglobulina humana venosa de 400 mg/kg peso. En los pacientes que han sido hospitalizados por infecciones severas, se recomienda una dosis de 400 mg/kg el día inicial, con una dosis igual extra a los 10 días; luego, se recomienda evaluar la necesidad de continuar con un programa regular de suministro de gamaglobulina venosa.
VI. Seguimiento de los pacientes
- Los pacientes con diagnóstico de deficiencia de las células fagocíticas se deben programar para un control mensual por consulta externa, evaluando su estado clínico y supervisando la evolución desde el punto de vista de laboratorio; estas citas serán también de utilidad para las labores de educación y prevención dirigidas al paciente y la familia.
- Se recomienda realizar anualmente un control de: pruebas de función pulmonar y endoscopia digestiva alta, pruebas de función hepática y renal, química sanguínea.
- Otros análisis de laboratorio están indicados cuando sean necesarios para el estudio de alguna enfermedad: directos y cultivos de secreciones; coprológico; estudios imagenológicos, etc.
Referencias
- 1. García de OD, Patiño PJ, Salgado H, López JA, Montoya CJ, Pérez JE. Evaluación del paciente con inmunodeficiencia. Síndrome de infección recurrente patológica. Medicina y Laboratorio 1997; 7: 545-575.
- 2. Paul ME, Shearer WT. Approach to the evaluation of the immunodeficient patient. In: Rich RR, Fleisher TA, Schwartz BD, eds. Clinical Immunolgy: Principles and Practice. St. Louis: Mosby 1996: 609-620.
- 3. Carneiro-Sampaio MMS, Grumach AS, Manissadjian A. Laboratory screening for the diagnosis of children with primary immunodeficiencies. J Invest Allergol Clin Imunol 1991; 1: 195-200.
- 4. Shearer WT, Paul ME, Smith CW, et al. Laboratory assesment of immune deficiency disorders. Immunol Allergy Clin North Am 1994; 14: 265-299.
- 5. López M, Fleisher T, de Shazo RD. Use and interpretation of diagnostic immunologic laboratory test. JAMA 1992; 268: 2970-2990.
- 6. Knutsen AP, Fischer TJ. Primary Immunodeficiency Diseases. En: Manual of Allergy and Immunology. Lawlor GJ, Fischer TJ, Adelman DC (eds). Little Brown and Company, Boston 1995; 397-424.
- 7. Stiehm ER. Conventional therapy of primary immunodeficiency diseases. En: Primary immunodeficiency diseases. Ochs HD, Smith CIE, Puck JM (eds). Oxford University Press, Oxford 1999; 448-458.
- 8. Polmar SH, Sorensen RU. Immunoglobulin replacement theraphy in primary immunodeficiency diseases. In: Rich RR, Fleisher TA, Schwartz BD, eds. Clinical Immunolgy: Principles and Practice. St. Louis: Mosby 1996: 1865-1873.
- 9.Zora JA, Silk HJ, Tinkelman DG. Evaluation of postimmunization pneumococcal titers in children with recurrent infections and normal levels of immunoglobulin. Ann Allergy 1993; 70: 283-287.
“Suele ser más fácil recuperarse de un fracaso que formarse a partir del éxito”
Michael Eisner