DISCUSION
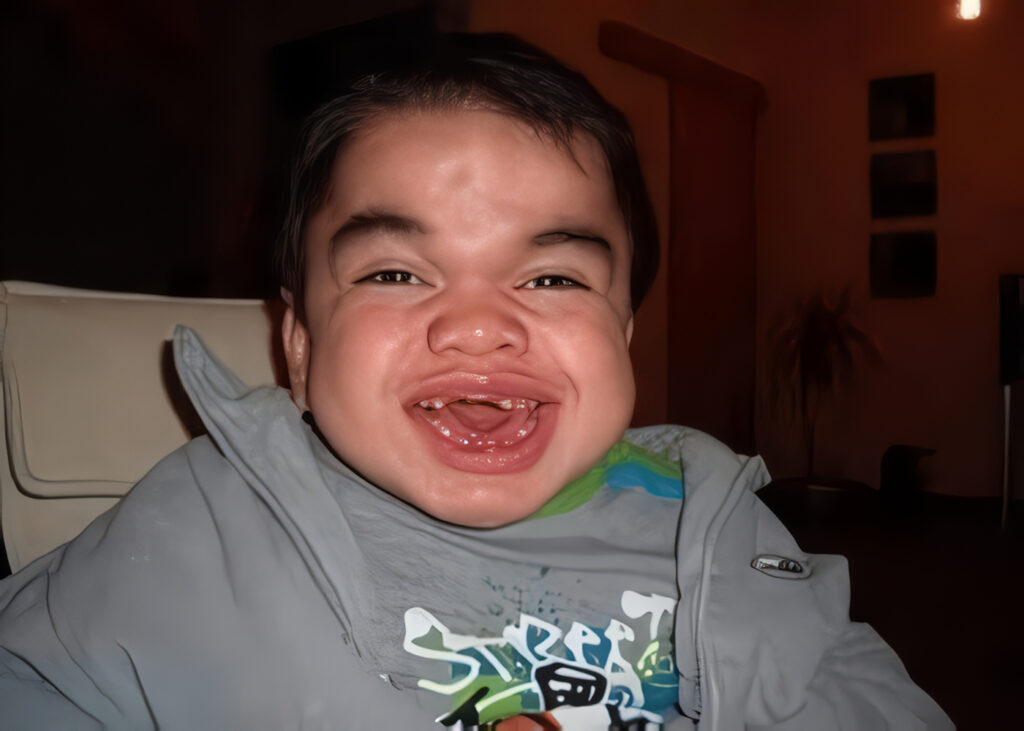
La Mucopolisacaridosis tipo VI, también conocida como síndrome de Maroteaux-Lamy, es una enfermedad metabólica hereditaria, de baja prevalencia, causada por el déficit de enzimas especificas como lo es la Arilsulfatasa B, implicada en el catabolismo de los glicosaminoglicanos (GAG); como consecuencia son acumulados en los lisosomas de las células de diferentes órganos, produciendo un daño progresivo y permanente a largo plazo, como se presento en el caso anteriormente nombrado, que presentó la sintomatología característica de la enfermedad y complicaciones secundarias a la misma. Es conveniente analizar la conservación aparente de la agudeza visual puesto que en muchos casos reportados de MPS tipo VI se presenta opacidad corneal lo que implica el trasplante de corneas, igualmente es recomendable extraer quirúrgicamente amígdalas y adenoides para mejorar la respiración de los individuos afectados por trastornos de las vías respiratorias y apnea del sueño, describir los hábitos de dormir con el fin de determinar el estado de las vías respiratorias y la posible necesidad de oxigeno durante la noche algunos pacientes pueden requerir la inserción quirúrgica de un tubo endotraqueal para ayudarlos a respirar.
Es posible realizar un diagnóstico prenatal usando técnicas de amniocentesis o vellosidades coriónicas para verificar si el feto tiene una copia del gen defectuoso o en su defecto las dos copias que implican el padecimiento del trastorno. Una correcta asesoría genética ayuda a los padres con antecedentes familiares de MPS tipo VI a determinar si son portadores del gen mutado causante de la alteración.
Los tratamientos disponibles son dirigidos a mejorar la calidad de vida de los pacientes, la evidencia científica disponible ofrece una visión amplia acerca de las diferentes pruebas de laboratorio empleadas para el diagnostico de este tipo de enfermedades, lo que permite hacer un diagnostico temprano para mejorar el pronóstico y minimizar el daño irreversible que causa la enfermedad. Cabe mencionar que en el caso del paciente no se ha presentado progresión de la enfermedad puesto que han disminuido las complicaciones propias de su enfermedad, desde el momento que se inicia manejo farmacológico con galsulfasa. Podemos asegurar que tras la administración de galsulfasa, a dosis de 20mg diluidos en 100cm3 de solución salina normal al 0.9%, 10cm3/hora la primera hora, luego 30cm3/hora lenta hasta terminar infusión (12, 14-16), el paciente mejoró notablemente, su resistencia física y su condición pulmonar, comparable con los resultados similares en respuesta al tratamiento con Galsulfasa, a dosis de 1mg/kg (9).
Los autores aseguran que no hay conflicto de interés alguno con el laboratorio productor de la Galsulfasa y además, que el escrito se hizo bajo consentimiento informado del paciente y sus familiares.
AGRADECIMIENTOS
De manera especial, por sus aportes y apoyo en el desarrollo del estudio, a los profesionales de la salud del Hospital Infantil Los Ángeles, así como a la Dra. Edilma Bastidas, Gerente Científica.
REFERENCIAS
1. Mabe P. Las mucopolisacaridosis. Rev Chil Nutric. 2003; 31(1): 8-16.
2. Natl Instit Neurol Disord Stroke. USA. Mucopolysaccharidosis and Mucolipidosis. 2007.
3. Wilcox WR. Lysosomal storage disorders: the need for better pediatric recognition and comprehensive care. J Pediatr. 2004; 144(5): 3-14.
4. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orph J Rare Dis. 2010; 5(5):1186-1750.
5. Litjens T, Baker EG, Beckmann KR, Morris CP, Hopwood JJ, Callen DF. Chromosomal localization of ARSB, the gene for human N-acetylgalactosamine-4-sulphatase. Human Gen 1989; 82(1): 67-68.
6. Karageorgos L, Brooks D. A, Pollard A, Melville E. L, Hein L. K, Clements R, et al. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Genome.net. 2007. Acceso: 3 de Julio 2012. Disponible en: https://www.genome.jp/dbget-bin/www_bget?omim+611542
7. Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics, 2007; 120 (2):405-18.
8. Hopwood JJ, Elliott H, Muller VJ, Saccone GT. Diagnosis of Maroteaux-Lamy syndrome by the use of radiolabelled oligosaccharides as substrates for the determination of arylsulphatase B activity. Biochem J. 1986; 234(3):507–514.
9. Swiedler SJ, Beck M, Bajbouj M, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with mucopolysaccharidosis VI (Maroteaux- Lamy syndrome). Am J Med Genet. 2005; 134(2): 144-150.
10. Rmatz P, Yu ZF, Giugliani R, Schwartz IV, Guffon N, Teles EL. Enzyme replacement therapy for mucopolysaccharidosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase, J Inherit Metab Dis. 2010; 33(1):51-60.
11. Correa Luz N, Mucopolisacaridosis. Precop SCP, Ascofame. 2004. Vol 1 30-36.
12. Rosselli D, Rueda JD, Solano M. Ethical and economic considerations of rare diseases in ethnic minorities: the case of mucopolysaccharidosis VI in Colombia. J Med Ethics 2012. (May 1st, published ahead of print).
13. A González, Meneses A, Barcia Ramírez, JL Díaz Rodríguez. Protocolo de actuación en las Mucopolisacaridosis. Protoc Diagn Ter Pediatr. 2010; 1: 24-36.
14. Selim T, Koseoglu, Harmatz P, Turbeville S, Bonito H. Reversed papilledema in an MPS VI patient with galsulfase (Naglazyme®) therapy. Int Ophthalmol. 2008; 29(4): 267-269.
15. Harmatz P. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis VI: clinical facts and figures. Turk J. Pediat 2010; 52: 443-449.
16. Harmatz P. Entering a new treatment age for mucopolysaccharidosis VI disease: a search for better markers of disease progression and response to treatment. J Pediatr (Rio J). 2008. 84:130–135.
Fecha de recibido: Mayo 15 de 2012
Fecha de aprobado: Octubre 5 de 2012
Direcció para correspondencia:
ivan.hernandez@ucc.edu.co