En 1994 en la conferencia de consenso de Chapel Hill, que fue una reunión de nomenclatura se definió a la GW como una vasculitis primaria y de acuerdo al tamaño de los vasos y a la inmunopatogénesis. La GW se define como una inflamación granulomatosa que compromete el tracto respiratorio alto y bajo cuya inflamación combina una inflamación necrotizante que afecta vasos de pequeño y mediano calibre y se asocia a glomerulonefritis necrotizante y anticuerpos anti-citoplasma del neutrófilo o C-ANCA.
Criterios clasificatorios se establecieron en 1990 por la American College of Rheumatology y posteriormente los de Chapel Hill en 1994, pero solo se aplican después de establecer el diagnóstico de la vasculitis sistémica, ya que en la fase inicial de la enfermedad puede rentringirse a la enfermedad granulomatosa del tracto respiratorio bajo en ausencia de vasculitis sistémica y la forma limitada descrita por Carrington y Liebow en 1966.
Personalmente he revisado todas las publicaciones sobre formas limitadas en tracto respiratorio alto, órbita, glándula mamaria, próstata, etc. y ningún artículo incluyendo el inicial ha evolucionado a una forma sistémica, además no reúnen los criterios clasificatorios y son patologías granulomatosas regional que se mejoran y además se curan rápidamente.
En mi criterio personal creo que existe la patología granulomatosa regional, pero no la forma limitada de GW.
La presencia de dos o más criterios tienen una sensibilidad del 88.2% y una especificidad del 92%.
Curso de la enfermedad
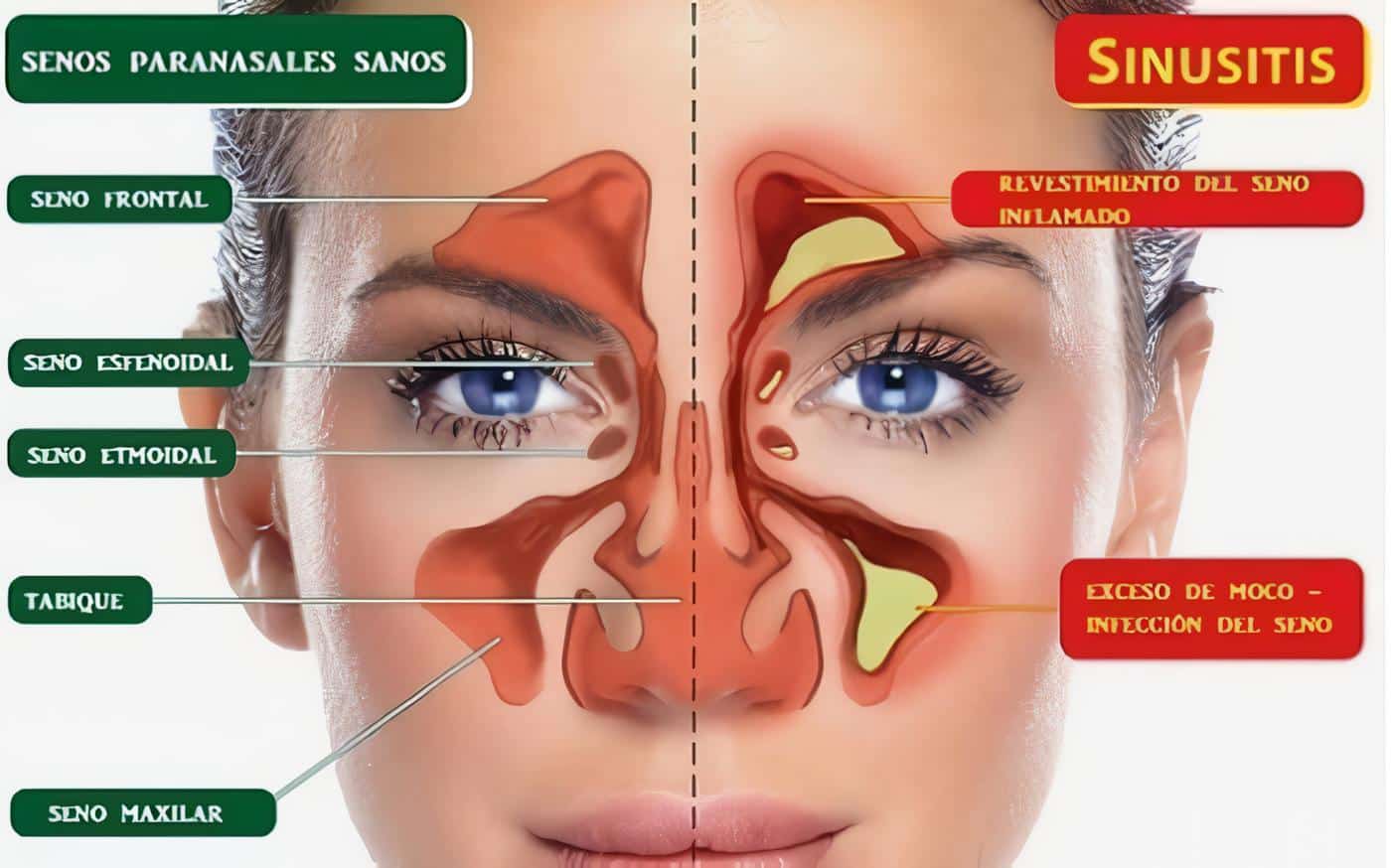
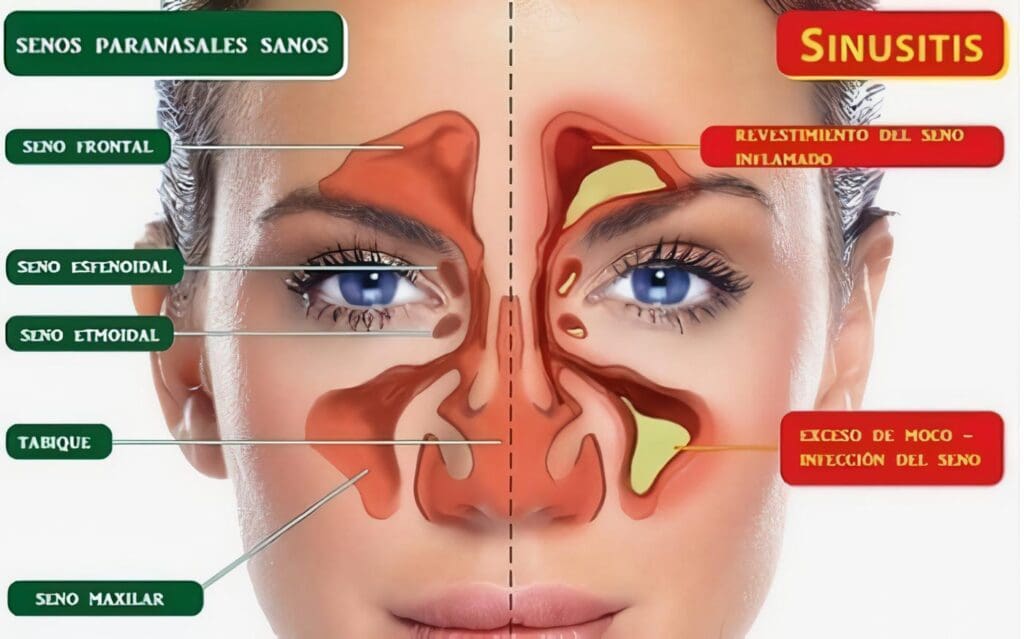
De acuerdo a la evolución de la enfermedad se pueden plantear dos etapas, una etapa inicial presumiblemente su mecanismo patogénico es de tipo celular y granulomatoso, predomina la etapa THI y se caracteriza por inflamación granulomatosa del tracto respiratorio alto, y se caracteriza por sinusitis seca, epistaxis, deformación en silla de montar, perforación del septum, otitis media y mastoiditis, las cuales por continuidad puede extenderse a la cavidad oral, faringe, tráquea, espacio retrobulbar, bronquios y menos frecuentemente a cerebro.
Las lesiones granulomatosas (tumefacciones o masas):
Se pueden observar a través de radiografía simple de senos paranasales, pero preferiblemente la resonancia magnética de estas áreas, la positividad del C-ANCA se encuentra en el 50% de los pacientes en esta etapa y la confirmación histológica es importante para el diagnóstico, pero muchas veces no se observa las lesiones granulomatosas y menos la vasculitis en biopsias de senos paranasales.
La transición a la forma generalizada o sistémica podría ser TH2 ya que predomina la respuesta inmunitaria humoral y aparece la vasculitis, se asocia a síntomas constitucionales como fiebre, sudoración nocturna y pérdida de peso.
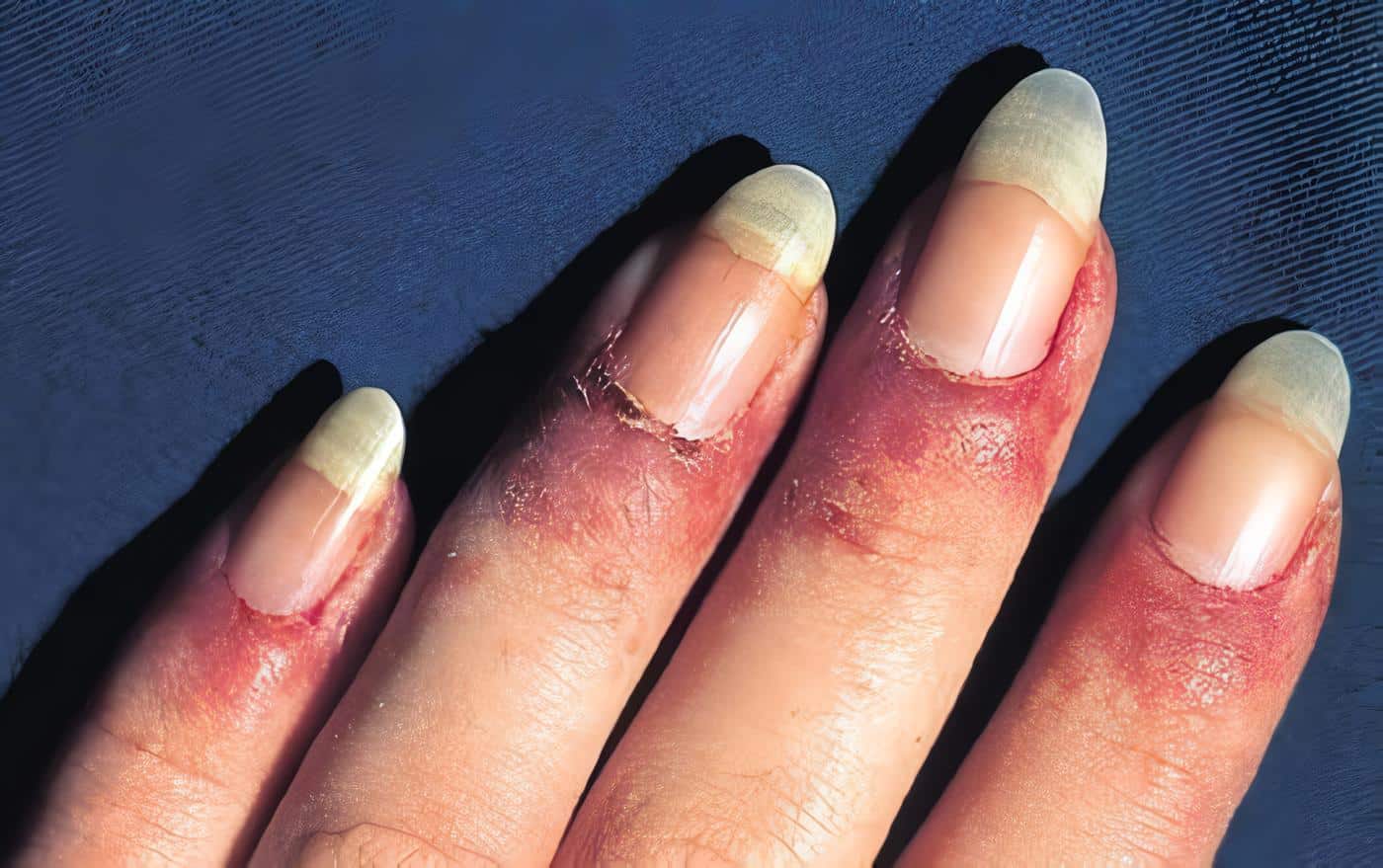
Además en esta fase se puede observar artralgias, artritis, úlcera maleolar, escleritis, proptosis ocular y en ocasiones lesiones de púrpura palpable por vasculitis leucocitoclástica, neuropatía periférica y compromiso renal, que suele ser o una capilaritis renal y evolucionar a una glomerulonefritis focal y segmentaria hasta una glomerulonefritis necrotizante difusa y/o glomerulonefritis rápidamente progresiva.
La forma agresiva o fulminante de GW se puede presentar en algunos casos sin pasar por la fase inicial o granulomatosa en el síndrome de pulmón – riñón, con hemorragia alveolar ó capilaritis y glomerulonefritis rápidamente progresiva. Se ha descrito un subgrupo de GW generalizada pero menos agresiva y C-ANCA negativo. Generalmente los pacientes con capilaritis pulmón y síndrome de pulmón – riñón son refractarios al tratamiento.
Criterios clasificatorios para la granulomatosis de Wegener GW 1990- (American College of Rheumatology)
")
En la fase de vasculitis de la enfermedad se puede encontrar el Ab contra la proteinasa – 3 ó C-ANCA en el 95% de los casos.
En esta fase de vasculitis se han descrito algunos casos con compromiso gastro – intestinal. El primer informe lo hizo Walton al describir 54 casos de GW y describe 13 pacientes con afección gastro – intestinal. En el artículo de Carrington y Liebow sobre “la forma limitada”, describen dos pacientes de 16 que tenían hemorragia a nivel de úlceras yeyunales.
En 1982 Mc Nabb y col encontraron un caso de GW, un paciente de 50 años con perforación del intestino delgado. Los pacientes con manifestaciones gastro – intestinales si no se diagnostica rápidamente se pueden encontrar casos refractarios al tratamiento.
Otros casos de vasculitis refractarias
Se puede presentar casos de vasculitis refractarias en aquellos casos que coexistan vasculitis y trombosis, a continuación enumeramos a través de tablas las enfermedades:
")

Síndromes paraneoplásicos
Las manifestaciones paraneoplásicas, los síndromes mieloproliferativos, los linfomas de células T pueden manifestarse con alteraciones cutáneas de tipo vasculítico (linfomonocítico ó leucocitoclástico ó infiltrados perivasculares) que simulan vasculitis. En estos casos se pueden presentar problemas refractarios al tratamiento, en ocasiones por la dificultad diagnóstica y en otros por las dificultades del tratamiento.
Vasculitis inducida por medicamento: En ocasiones los casos refractarios se presentan porque no se piensa en esta patología.
Medicamentos asociados a vasculitis.
I. Factores de crecimiento hematopoietico
a. Factor estimulante de colonias de granulocitos
b. Factor estimulante de colonias de granulocito – macrófago
II. Interferones
a. Interferón a
b. Interferón b
c. Interferón g
III. Vasculitis – ANCA positivos
a. Hidralazina
b. Medicamentos antitiroideos
1. Propiltiouracilo
2. Metimazole
3. Carbimazole
IV. Miscelánea
a. Minociclina
b. Penicilamina
Tratamiento de las vasculitis refractaria
En 1958 Walton publicó su artículo sobre GW y en esa época la mortalidad por esta patología al primer año era del 80%, tres décadas después Hoffman sobre un estudio de Cohorte de 158 pacientes reduce la mortalidad a un 13 % en un período de siete años, al utilizar el esquema de Fauci con la ciclofosfamida por vía oral asociada a los corticosteroides, así de esta forma se puede producir una remisión en el 90% de los pacientes con esta patología; además utilizando otros medicamentos alternos como el metrotexate, la azotioprina y la ciclosporina. La azotioprina es un buen medicamento de mantenimiento en aquellos casos con vasculitis refractaria con función renal alterada.
No existen muchas opciones terapéuticas para los casos que hemos analizado de GW donde existen problemas relacionados con resistencia al tratamiento, para ello además de los medicamentos mencionados se puede utilizar alguna combinación de medicamento de acuerdo al tipo de vasculitis y el órgano blanco comprometido. Si estas alternativas no funcionan es importante la toma de una decisión rápida y para ello se puede utilizar otros medicamentos tales como: inmunoglobulinas por vía IV (400 mg/kg IV x 5 días).
Este esquema además de utilizarse para la enfermedad de Kawasaki, se ha utilizado en pacientes con GW y la poliangeitis microcópica por Jayne y Lockwood; en todos los pacientes en los que se utilizó la IVIG, II en total hubo un beneficio sustancial, con reducción de la inmunosupresión después de un seguimiento de 12 meses. Hubo una reducción de los títulos de ANCA en el 50%. En un abstracto de la ACA (American College of Rheumatology) presentado en 1994 Richter y col al utilizar IVIg en vasculitis asociada a ANCA de tipo refractario solo respondieron el 40%, es decir hubo una pobre respuesta.
Anticuerpos monoclonales
Mathieson y col en 1990 utilizan anticuerpos monoclonales contra algunos receptores blancos de las células T, específicamente contra CD4 y CD52, utilizando para ello una terapia secuencial por vía intravenosa. Pienso que este tipo de terapia biológica se encuentra en una fase experimental y no existen resultados adecuados para implementarla para las vasculitis refractarias, a pesar del estudio de Lockwood y col quienes la utilizaron en cuatro pacientes con vasculitis refractaria con resultados aceptables.
Globulina anti-Timocito
Los anticuerpos anti-Timocito policlonales han sido utilizados en cuatro pacientes con GW por Hagen y col con resultados aceptables.
Plasmaféresis
Actualmente no existe evidencias que soporten la prescripción de la plasmaféresis en pacientes con PAN asociado o no a hepatitis B ó C, Pusey y col, Gillevin y col, Madore y col plantean la utilización de la plasmaféresis en pacientes con glomerulonefritis proliferativas como causa de una insuficiencia renal (creatinina > 500 U moles/litro), en estos casos la plasmaféresis puede mejorar la función renal y detener los procedimientos de diálisis. Actualmente se realiza un estudio controlado para este protocolo.
Tratamiento de la hepatitis B y otras infecciones virales asociadas a la PAN
A pesar de utilizar los corticosteriodes y los inmunosupresores en los pacientes con hepatitis B (HBV) asociada a PAN logrando beneficio a nivel de la vasculitis, estos medicamentos pueden perpetuar el proceso crónico de la HBV y facilitar el desarrollo de la cirrosis y finalmente complicarse con un carcinoma hepatocelular, por ello es fundamental utilizar agentes anti-virales como la vidarabina (Vira – A) o el interferón alfa – 2b. Guillevin y col han tratado aproximadamente a 45 pacientes con esta asociación de HBV y PAN (35 con Vira – A y 6 con IFN a2b, y la tasa de sobrevida en 10 años ha sido del 83%.
La tasa de seroconversión (HBeAg / HBe Ab) se observó en 21 de los 41 pacientes es decir en el 24.4%. Estos resultados son mejores que los obtenidos con esteroides asociado o no con ciclofosfamida y utilizando plasmaferesis, ya que la seroconversión en estos casos fue bastante rara.
Factores pronósticos para escoger el tratamiento de las vasculitis graves y de las vasculitis resistentes.
Dos grupos de origen europeo han introducido la evaluación cuantitativa de la actividad de las vasculitis a través del Birminghan vasculitis Activity Score (BVAS) ideado por Lugmani y Bacon que ha sido validado en múltiples países y por nuestro grupo en el Hospital San Juan de Dios de Bogotá y la versión modificada del B.V.A.S., liderada por Bacon, que está en investigación en Europa llamada Vital (vasculitis integrated total Assessment log), a través de estudios multicéntricos en Europa denominada ECSYSVASTRIAL (European multicenter therapeutic vasculitis trial) y a través del índice de extensión de la enfermedad denominado DEI (Disease Extend Index) liderado por Groot, Gross, Reinhold – Keller en Alemania. A través de estos índices analizamos el estado del compromiso de los órganos afectados y podemos racionalizar el esquema del NIH (National Institute of Health), la fase de remisión o establecer si hay resistencia a la terapia.
Otros indicadores utilizados para analizar la gravedad de las vasculitis es propuesto por el grupo francés de Guillevin y el de Lugmani y Bacon denominado five factors score (FFS) que demuestra un gran valor pronóstico y de acuerdo a los parámetros utilizados que tiene el paciente se puede analizar la gravedad.
Estos cinco factores son:
1. Proteinuria mayor de 1.0 gr/día
2. Insuficiencia renal (creatinina > 140 umoles/litro)
3. Cardiomiopatía
4. Alteraciones gastro – intestinales
5. Compromiso del sistema nervioso central. Cuando el índice FFS es 0, la mortalidad a cinco años es del 12%, si el índice es FFS I, la mortalidad se incrementa al 26% y si es 2 la mortalidad es del 46%. Si el índice es de 5, la mortalidad es mayor del 90%. Todos estos índices deben ser utilizados por nuestros reumatólogos para de esta manera analizar el compromiso del órgano y el pronóstico de nuestros pacientes.
Lecturas recomendadas
- 1. Iglesias – Gamarra A, Méndez O, Valle R, Osorio E. Vasculitis Necrotizantes y síndromes asociados. Salvat ed. 1982
- 2. Iglesias – Gamarra A, Valle R, Igea E, Vásquez G, Salázar M. Análisis histórico de las vasculitis, su clasificación y propuesta para su entendimiento. Biomédica 1993; 18: 40.
- 3. Lhote F, Cohen P, Guillevin L. Polyarteritis nodosa, microscopic polyangiitis and Churg – Strauss Syndrome. Lupus 1998; 7: 238.
- 4. Dillon MJ. Childhood Vasculitis. Lupus 1998; 7: 259.
- 5. Hunder GG. Giant cell arteritis. Lupus 1998; 7: 266.
- 6. D’Cruz D. Vasculitis in systemic lupus erythematosis. Lupus 1998; 7: 270.
- 7. Ferri C, Civita Lla, Longobardo G, Zignego AL, Pasero G. Mixed cryoglobulinemia: a cross- road between autoinmune and lymphroproferative disorders. Lupus 1998; 7: 275.
- 8. Kallenberg CGM, Heeringa P. Pathogenesis of vasculitis Lupus 1998; 7: 280.
- 9. De Groot K, Gross WL. Wegener’s granulomatosis: disease course, assessment of activity and extent and treatment. Lupus 1998; 7: 285.
- 10. Hoffman GS. Vasculitic Syndromes: Editorial Overview. Curr opinion in Rheumatology 1998; 10: 1.
- 11. Nowack R, Flores – Suárez LF, Vander Woude FJ. New developments in pathogenesis of systemic vasculitis. Curr Opinion in Rheumatol 1998; 10: 3.
- 12. Cid MC, Font C, Coll – Vinent B, Grau JM. Large vessel vasculitides. Curr Opinion in Rheumatol 1998; 10: 18.
- 13. Merkel PA. Drug Associated with vasculitis. Lupus 1998; 16: 45.
- 14. Mandel BF, Calabrese LH. Infection and systemic vasculitis Lupus 1998; 10: 51.
- 15. Danning CL, Illei GG, Boumpas DT. Vasculitis associated with primary rheumatologic diseases. Lupus 1998; 10: 58.
- 16. López – Quijano JM, Torre – Bouscoulet LT, Magaña – Aguino M, Abud – Mendoza C. Anticuerpos antineutrófilo: inmunoterapia, enfermedades asociadas y esquemas de tratamiento. Rev. Mexicana de Reumatología 1998; 13: 150.
Referencias Recomendadas
- 17. Cotch MF, Rao JK. New insights into the epidemiology of systemic vasculitis. Current Opinion in Rheumatol 1996; 8: 19.
- 18. Jennette JC, Falk RJ, Andrassy K. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994; 37: 187.
- 19. Walton EW. Giant cell granuloma of the respiratoy tract (Wegener’s granulomatosis) BMJ 1958; 265-270.
- 20. Hoffman GS, et al. Wegener’s granulomatosis. Analysis of 158 patients. Ann Intern Med 1992; 116: 488
21. Guillevin L, Lhote F. Treatmentof polyarteritis nodosa and microscopic poliangiitis. Arthritis Rheum 1998; 41: 2100 - 22. Jayne DR, Lockwood CM. Intravenous inmunoglobulin as sole therapy for systemic vasculitis Br J Rheumatol 1996; 35: 1150.
- 23. Richter C, Schnabel A, et al. Treatment of ANCA – associated systemic vasculitis with high-dose intravenous inmunoglobulin (Abstract). Arthritis Rheum 1994; 37 suppl 9: 9353
- 24. Mathieson P. Cobbold S, Hale G, et al. Monoclonal – antibody therapy in systemic vasculitis. N. Engl J Med 1990; 232: 250.
- 25. Lockwood CM, et al. Long – term remission of intractable systemic vasculitis with monoclonal antibody therapy. Lancet 1993; 341: 1620.
- 26. Hagen EC et al. Compassionate treatment of Wegener’s granulomatosis with rabbit anti – thymocyteglobulin Clin Nephrol 1995; 43: 351.
- 27. Pusey CD, Gaskin G, Redd AJ, Treatment of primary systemic vasculitis APMIS Suppl 1990; 19: 48
- 28. Guillevin L. Cevallos R. Et al. Treatment of glomerulonephritis in microscopic polyangiitis and Churg – Strauss syndrome. Indications of plasma exchanges. Metaanalysis of 2 randomized studies on 140 patients 32 with glomerulonephritis. Ann Med Interne. París 1997; 148: 198.
- 29. Madore F, lazarus M. Brady H. Therapeutic plasma exchange in renal diseases. J. AM. Soc Nephrol 1996; 7: 367.
Bibliografías Recomendadas
- 30. Guillevin L. Lhote F. Et al. Polyarteritis nodosa related to hepatitis B virus: a prospective study with long – term observation of 41 patients. Medicine (Baltimore) 1995; 74: 238.
- 31. Lugmani RA, et al. Birminghan Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis QJ Med 1994; 87: 671.
- 32. Bacon PA, et al. Vital assessment of vasculitis. Clin Exp Rheumatol 1995; 13: 275.
- 33. Guillevin L. Lhote F, et al. Prognostic factors in polyarteritis nodosa and Churg – Strauss syndrome: a prospective study in 342 patients. Medicine (Baltimore) 1996; 75: 17.
- 34. Lugmani R, Exley A, et al. Disease assessment and management of the vasculitides. Baillieres Clin Rheumatol 1997; 11: 423.