Los tumores endocrinos gastroenteropancreáticos secretan hormonas activas y pueden estar asociados con síndromes clínicos que los distinguen. Hasta la fecha se han descrito. por lo menos, ocho síndromes clínicos diferentes relacionados con tumores funcionantes de los islotes del páncreas, y las hormonas responsables han sido identificadas: insulina (insulinoma), gastrina (gastrinoma o síndrome de Zollinger-Ellisonl. péptido intestinal vasoactivo o YIP (síndrome WDHA), glucagón (glucagonoma), ACTH (síndrome de ACTH ectópico, hormona paratiroidea o PTH (hiperparatiroidismo ectópico) y 5-hidroxitriptamina (síndrome carcinoide) (12). Es posible que con el transcurso del tiempo, un mismo tumor sea capaz de secretar diferentes hormonas en forma secuencial. En cada instancia, las manifestaciones clínicas principales del tumor son debidas a la liberación excesiva, no controlada, de péptidos hormonales en la circulación. Estas hormonas se producen naturalmente y son importantes en la mediación de muchos procesos. Sin embargo, en pacientes con tumores pancreáticos endocrinos, la liberación y secreción de estas hormonas no están bajo regulación fisiológica normal. Debido a los extremadamente altos niveles de hormonas, se desarrollan signos y síntomas característicos (Tabla 1). La colección de células neuroendocrinas difusas gastroenteropancreáticas es un gran reservorio que secreta ami nas y péptidos con el propósito de regular y modular el control fisiológico normal del metabolismo de carbohidratos, la digestión de proteínas y grasas y la provisión de un pH ácido o alcalino, apropiado para la asimilación de nutrientes. Estas células neuroendocrinas ejercen su influencia fisiológica por medio de una actividad paracrina sobre las células vecinas, una actividad neurocrina a través de neurotransmisores aminérgicos y peptidérgicos y por su tradicional función endocrina (humoral).
Existe alguna confusión en cuanto a la nomenclatura con que han sido denominados los tumores que surgen de estas células. Términos como argentafinomas, tumores de células enterocromafínicas, tumores neuroendocrinos, tumores carcinoides y APUDomas, han sido comúnmente usados para clasificarlos. Revisemos en una breve historia, el conocimiento actual de la estructura y función de la célula neuroendocrina gastroenteropancreática.
En 1870 se reconoció una población de células con un citoplasma granular distinto al de otros elementos del epitelio mucoso del tracto gastrointestinal (13). Estas células fueron llamadas de Kultschitzky por la creencia errónea de que ellas fueron observadas por primera vez por Kultschitzkyen 1897 (14). Por sus similitudes morfológicas, se pensó que representaban una población homogénea y por su recuerdo cercano a las células cromafínicas de la médula adrenal, se les designó como células enterocromafínicas (15). Su naturaleza endocrina fue propuesta por primera vez en 1906 (16) Y posteriormente se sugirió que los mafínicas (16). Otros investigadores observaron (17) que tanto las células de los tumores carcinoides como las enterocromafínicas. tenían una afinidad por las sales de plata (argentafinidad). Más tarde se encontró que si las secciones histológicas eran tratadas con una sustancia reductora antes de ser expuestas a las sales de plata. un gran número de gránulos celulares pueden ser demostrados. Las células coloreadas después de tal tratamiento fueron designadas como células argirófilas.

Ya desde 1938, se postuló que un grupo de células distribuidas a todo lo largo del tracto gastrointestinal, pero preferentemente en la parte distal (íleon, apéndice, duodeno). con características de citoplasma claro y afinidad distinta por las sales de plata (reactividad argentafín y argirófila). constituían un “sistema epitelial endocrino difuso”, y les fue asignada una función paracrina (18). Más tarde, en 1968. se reconoció que estas células tenían características citoquímicas y estructurales similares, ya que ellas contenían ami nas en su citoplasma y eran capaces de captar precursores de ami nas y usaban una enzima descarboxilante para convertir estos precursores en aminas y péptidos o en ambos. que almacenaban en gránulos secretorios dentro del citoplasma (19). Se admitió también que esta habilidad por la captación de precursores de minas y descarboxilación (APUD). era compartida por otras células a lo largo del cuerpo. La teoría en boga sostenía que estas y otras células similares eran de origen neuroectodérmico, lo cual implica que ellas derivan de un mismo tejido embrionario (cresta neural) y constituyen un sistema neuroendocrinobdifuso y que actúan en concierto con el sistema nervioso autónomo para controlar las funciones de todo el aparato gastrointestinal (20). Se basó la teoría en la opinión de que estas células se diferenciaban de una célula progenitora común y que algunos aspectos metabólicos eran retenidos para poder entonces ser usados en la identificación de esta relación. Existen numerosos marcadores neuronales compartidos por las células APUD (14, 21); por ejemplo. a varios de ellos encontrados en las células gastroenteropancreáticas neuroendocrinas y en las neuronas, son la enolasa neurón específica, una acetilcolinesterasa específica, sinaptofisina, receptores del toxoide tetánico, proteína S-lOO y cromograninas A, B, C. Sin embargo. características compartidas por las cél ulas neurales y las APUD no prueban un origen común, ya que esta hipótesis ha sido revisada por recientes decubrimientos en el área de la biología celular y citoquímica, que han puesto en duda los conceptos originales. El término APUD se considera ahora inadecuado porque varios tipos de células incluidas en el sistema, no metabolizan aminas (22). Además. existen ahora evidencias que sugieren que algunas células de tipo APUD, en particular las neuroendocrinas gastroenteropancreáticas, no se originan en la cresta neural, pero sí en el endodermo (21,23). Se ha insinuado, por lo tanto, que existen dos sistemas relacionados, los cuales incluyen más de 40 tipos de células (22), el sistema endocrino endodérmico difuso. que consta de células epiteliales de origen endodérmico y el sistema neuroendocrino difuso que comprende células que se originan en la cresta neural (24).
Sin embargo. se mantiene la posibilidad de que exista una célula precursora ectoblástica común para todas las células endocrinas (21. 25). Este precursor puede ser implantado en todas las capas germinales durante la granulación (26).
Después de la unificación de las ciencias básicas y los principios clínicos por el concepto APDO. los tumores que se originaron de estas células. fueron llamados apudomas. Aunque nuevas evidencias han establecido que las células del sistema gastroenteropancreático no derivan de la cresta neural y, por lo tanto, la teoría APDO puede no sostenerse por lo del origen común de todas las células APDO, esta teoría. no obstante. ha sido responsable para la apreciación de que las células endocrinas en el tracto gastrointestinal. en las adrenales. en las paratiroides y otros órganos endocrinos, tienen predeterminación neuroectodérmica y características como aquellas de las neuronas del sistema nervioso central. así como también las del sistema simpático y parasimpático. La palabra “apudoma”, indica un tumor que surge de células con características APDO, y está ahora firmemente establecida en el vocabulario médico. El concepto APUD también ha suministrado bases conceptuales para entender la ocurrencia de síndromes de neoplasias endocrinas múltiples (MEN), así como también, el potencial para que un tumor esté compuesto de varios tipos de células y secrete más de una hormona (27). La mayoría de los tumores neuroendocrinos hoy son designados por su producto humoral predominante.
Tanto como 19 tipos de células se consideran ahora que forman parte del sistema epitelial endocrino difuso gastroenteropancreático (13) Y casi 40 diferentes especies de sustancias farmacológicamente activas. péptidos y aminas biogénicas han sido identificadas en estas células (28). Por lo tanto, tumores que surgen de estas células constituyen un grupo heterogéneo, que aunque están estrechamente relacionados en términos de su mecanismo biosintético. así como en sus aspectos ultraestructural e histoquímico. demuestran diferencias claras en factores morfológicos. actividad endocrina y pronóstico. Funcionalmente, las neoplasias endocrinas gastroenteropancreáticas pueden ser entópicas (ortoendocrinas), con producción de cantidades anormales de su inherente hormona nativa u hormonas ectópicas (paraendocrinas), con producción de sustancias que se espera no deriven de aquellos tejidos, o no funcionantes. no asociadas con un síndrome clínico reconocible a causa de la liberación de péptidos en la circulación a pesar de la presencia de células endocrinas en el examen histológico. En general, tumores que producen hormonas entópicas son menos malignos que aquellos que las producen ectópicas. sugiriendo con ello. que las células tumorales que producen hormonas inapropiadas son más profundamente transformadas y más agresivas (29). Aunque los tumores endocrinos gastroenteropancreáticos varían en su potencial de malignidad, la extirpación quirúrgica después de su exacto diagnóstico y localización, sigue siendo la única forma curativa como terapia disponible. Las metástasis a los ganglios linfáticos deben ser extirpadas radicalmente. y si la masa tumoral es muy grande. ésta debe ser resecada en su mayor parte, cuando la extirpación completa no es posible, auncuando haya metástasis a distancia. El tratamiento médico de los tumores irresecables o residuales, consiste en medidas antisecretoras, terapia antihormonal o inhibición quimioterapéutica del crecimiento del tumor.
Insulinoma
El páncreas es una glándula con dos componentes. uno exocrina y otro endocrino. La porción exocrina es la más voluminosa y tiene un aspecto semejante al de una glándula salival; la parte endocrina está constituida por aproximadamente un millón de islotes de Langerhans homogéneamente distribuidos en el parénquima glandular. En términos generales miden entre 100 Y 200 micras de diámetro y poseen una rica red capilar. Una característica típica de la mayoría de los tumores de los islotes es su hipervascularización, lo que permite identificarlos con precisión en las angiografías.
Se han reconocido tres tipos de células en los islotes pancreáticos del humano; una célula alfa. elabora glucagón; la célula beta responsable de la secreción de insulina, y la célula delta que, en condiciones específicas, produce gastrina.
Los insulinomas son los tumores funcionantes de las células beta que son relativamente raros. El hiperinsulinismo endógeno fue el primer síndrome indentificado de excesiva secreción de hormona pancreática. El síndrome insulinoma se caracteriza por hipoglicemia sintomática. secundaria a hiperinsulinemia persistente. absoluta o relativa. La primera descripción de un tumor inductor a hiperinsulinismo viene de la Clínica Mayo en 1927. El tumor era un insulinoma maligno con múltiples metástasis hepáticas (30). Roscoe Graham, de Toronto, tiene el crédito de la primera extirpación quirúrgica exitosa de un insulinoma en 1929(31).
En 1922, Banting y Best aislaron una sustancia del páncreas que inducía a hipoglicemia y que llamaron insulina. Evarts Graham del Hospital Barnes de San Luis, enucleó exitosamente un insulinoma minúsculo (5 mm) en 1931. Este procedimiento fue idéntico a las enucleaciones que se efectúan hoy para el tratamiento de estos tumores.
El insulinoma es un tumor raro; algunos datos estadísticos señalan una incidencia de 4/5 millones de personas. El 6(Y/r) se presenta en hombres. Es poco frecuente en menores de 20 años y sólo el 20% está por debajo de 40 años, mientras que otro 40% está por encima de la sexta década de la vida. En el 90(k ocurre un tumor único y. cuando son múltiples. casi siempre forma parte del síndrome de neoplasia endocrina múltiple tipo I (MEN 1), esto es. tumores endocrinos en hipófisis, paratiroides y páncreas.
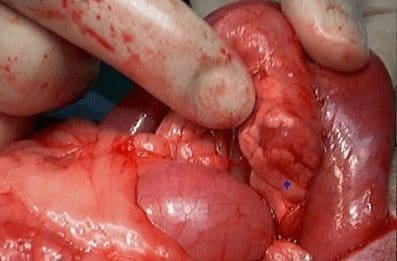
Pacientes a quienes se les encuentren múltiples tumores. deben ser evaluados para otras endocrinopatías. Son generalmente pequeños « 2 cm), benignos (el 90c¡¡.). uniformemente distribuidos en todo el páncreas (Fig. 1). Los tumores malignos tienden a ser más grandes, con un promedio de 6.2 cm de diámetro y pueden exhibir invasÍón capsular y de los vasos. Histológicamente es imposible diferenciarlos de los benignos. La presencia de metástasis a distancia ocurre en primera instancia en el hígado.

Los insulinomas son característicamente intrapancreáticos, firmes, encapsulados, de color rojizo marrón. Histológicamente hay dos tipos de insulinomas (32). Un tipo está caracterizado por células beta muy abundantes, bien granuladas, con insulina uniformemente inmunofluorescente, organizadas en un patrón trabecular y bajo contenido de proinsulina. El otro tipo, tiene escasas células beta, bien granuladas, con insulina irregularmente inmunofluorescente, con un patrón medular y alto contenido de proinsulina. Los insulinomas ectópicos son raros (2 a 3%). Se encuentran generalmente en la mucosa duodenal, el hilio esplénico o el ligamento gastroepiploico.
En los adultos con enfermedad de las células beta de los islotes pancreáticos e hiperinsulinismo, puede ser reconocido un número de entidades patológicas, tales como las que se mencionan a continuación:
l. Adenoma solitario
2. Adenomatosis
Macroadenomas solos
Microadenomas + microadenomatosis
Macroadenomas + hiperplasia
3. Nesidioblastosis
4. Hiperplasia
5. Carcinoma
La adenomatosis consiste en I ó múltiples macroadenomas junto con microadenomas intercalados entre islotes normales. Ocurre entre un 5 a 15% de los pacientes con hiperinsulinismo, aunque raramente se discute en la literatura. Los carcinomas (aproximadamente 5% de los pacientes con hiperinsulinismo orgánico) sólo pueden ser diagnosticados con certeza cuando están presentes metastásis regionales o a distancia.
La nesidioblastosis (neoproliferación difusa y diseminada de células productoras de insulina a partir de los conductos pancreáticos), es la principal causa de hiperinsulinemia en recién nacidos e infantes, reconocida en el pasado (33). Recientemente se le ha observado también en adolescentes y adultos (34-37); ocurre en aproximadamente el 5% de los pacientes con hiperinslinismo. Puede ser difusa o limitada incialmente a una parte del páncreas. La hiperplasia difusa de las células beta (proliferación excesiva y difusa de las células beta en los islotes de Langerhans) se ha informado en adultos. Stefanim y col (38) señalaron que de un total de 1.137 casos de hiperinsulinismo orgánico, 1.067 (93.8%) eran causados por insulinomas, y 70 (6.2%), por enfermedad difusa de las células de los islotes. Algunos pacientes asignados en la literatura como portadores de hiperplasia, pueden ser clasificados hoy más propiamente como nesidioblastosis, si se aplican técnicas apropiadas de coloración con inmunofluorescencia. La nesidioblastosis, la hipertrofia de los islotes, la hiperplasia, la adenomatosis y el adenoma, pueden ser la expresión de un mismo defecto básico llamado por Gabbay (39) “síndrome de desmaduración de las células de los islotes”, y “displasia celular endocrina” por Jaffe y col (40). El reconocimiento de microadenomatosis, nesidioblastosis o hiperplasia, es importante desde el punto de vista terapéutico, ya que es necesaria la remoción de una gran porción d~1 páncreas para el alivio del hiperinsulinismo y la hipoglicemia. En algunos pacientes, un 70 a 85% de pancreatectomía, logrará la cura; en otros, es esencial la terapia médica o la remoción adicional del tejido pancreático.