J. DE LA HOZ, MD, SCC.
Palabras claves: Glúndulas suprarrenales, Feocromocitoma, Hipertensión arterial, Catecolaminas, Bloqueo adrenérgico.
A pesar de su relativa infrecuencia. los feocromocitomas constituyen un grupo de causas de hipertensión arterial. potencialmente curable por la extirpación quirúrgica del tumor, siendo su etiologia letal. La apropiada utilización de modernas técnicas quimicas de laboratorio c!inico y de sofisticados métodos de imágenes diagnósticas, le permiten al clinico que sospecha su presencia. establecer o no el diagnóstico y localización de un tumor secretor de catecolaminas.
El tratamiento del feocromocitoma sigue siendo quirurgico: sin embargo. la atención estricta a los detalles preoperatorios es crucial si se quiere reducir la mortalidad. El uso juicioso de agentes de bloqueo adrenérgico y medidas para asegurar un volumen sanguíneo circulante normal. son de suma importancia en el periodo preoperatorio. Una completa y meticulosa exploración de toda la cavidad abdominal y de ambos lechos adrenales. es esencial en todos los pacientes con feocromocitoma. La monitoria hemodinámica y electrocardiográfica es importante la cirugia y el postoperatorio. ya que los cambios en la estabilidad cardiovascular pueden ser rápidos y dramáticos. Deben tomarse medidas apropiadas para revertir tales cambios. pero deben estar basadas en el completo entendimiento de los efectos de las catecolaminas. de las drogas que alteran estos efectos, de los problemas potenciales que el paciente con feocromocitoma puede experimentar.
Introducción
Ninguna de las causas endocrinas de hipertensión arterial es tan fascinante y desafiante para el clínico como lo es la producida por el feocromocitoma. Sus manifestaciones proteiformes pueden hacer el diagnóstico difícil y sus implicaciones pronósticas siniestras demandan un pronto reconocimiento y un experto manejo. El diagnóstico depende de la sospecha clínica, de la demostración de niveles altos de catecolaminas libres en el plasma o en la orina, o de la exacta localización del tumor por técnicas apropiadas de imúgenes que incluyen la TAC, la resonancia magnética y la escintigrafía con metayodobenzilguanidina I 131
Doctor Jaime De la Hoz. Prof Emérito de la Fac. de Med. de la U. Nal., Expresidente de la Sociedad Colombiana de Cirugia, Bogotá. D. c., Colombia.
(MIBG). La extirpación quirúrgica es el tratamiento de elección, a menos que el riesgo de la intervención sea abrumador o las metústasis a distancia ya hayan ocurrido en el momento de la toma de decisiones. El éxito final demanda una participación multidisciplinaria que aproveche las ventajas de la experiencia, habilidad y competencia de cirujanos, anestesiólogos e internistas.
Material y métodos
Un total de 13 pacientes operados para feocromocitomas han sido coleccionados y estudiados retrospectivamente. Unos han sido operados en el Hospital de San Juan de Dios y otros en clínicas privadas, en un período de 12 años. Ocho hombres y 5 mujeres. La edad promedio fue de 38 años. Todos tuvieron comprobación histológica de tumor cromafínico.
Ocho tumores ocurrieron en la suprarrenal derecha y 3 en la izquierda. La totalidad de éstos se clasificó como esporádicos. Ninguno formó parte del síndrome de neoplasia múltiple tipo MEN n. Doce fueron clasificados como benignos y l como maligno por su tamaño (más de 5 cm) y posterior presencia de metástasis hepática. Once estaban localizados en las glándulas suprarrenales, 1 paraórtico, por debajo del nacimiento de la mesentérica inferior feocromocitoma extraadrenal, órgano de Zuckerkandl), y l se clasificó como paraganglioma torácico. No hubo mortalidad operatoria. Las cifras tensionales se normalizaron, con excepción del caso del tumor maligno que mejoró por 6 meses, momento en que se detectaron metástasis hepáticas. Las crisis hipertensivas fueron controladas con metirosina, pero al ser suspendida ésta, el paciente falleció por accidente cerebrovascular.
Discusión
Desde el primer diagnóstico hecho por Labbe, Tinel y Doumer en 1922, y de la primera intervención quirúrgica exitosa lograda en 1926 por Cesar Roux y Charles Mayo, el feocromocitoma ha sido definido como un tumor benigno, intraadrenal, secretor de catecolaminas y, por lo tanto, un tumor hipertensivo. En las décadas siguientes se hizo evidente que la definición anterior no incluía todos los aspectos de los feocromocitomas. En realidad estos tu mores no son siempre intraadrenales; son, en ocasiones, ectópicos, bilaterales y pueden ocurrir con características genéticas. Un seguimiento a largo plazo de pacientes operados de un tumor, aparentemente benigno, puede descubrir metástasis tardías.
En los últimos 30 años ha crecido el número de síndromes hipertensivos claramente relacionados con la superproducción de ciertas hormonas. La mayor parte de ellos pueden curar con el tratamiento quirúrgico. Se incluyen en esta lista, el hiperaldosteronismo primario, el feocromocitoma, el síndrome de Cushing por adenoma o carcinoma de la corteza adrenal, la hiperplasia adrenocortical (adenoma o microadenoma hipofisiario, enfermedad de Cushing), la debida a tumores ectópicos productores de ACTH o la hiperplasia adrenocortical congénita por excesiva producción de desoxicorticosterona a consecuencia de deficiente producción de 11 beta o de 17 alfa hidroxilasa y el reninismo primario (tumores productores de renina). La hipertensión renovascular generalmente se considera una hipertesión endocrina, a la luz de la evidencia de que la renina-angiotensina tiene qué ver con su patogénesis. Complementan esta lista la hipertensión relacionada con el hipotiroidismo y los recientes trabajos pioneros de Dluhy y col, y de Walker y Edwards, los cuales han proporcionado nuevas y pertinentes guías para entender y corregir la biología y la fisiopatología de la hipertensión que se deriva del ’11- dosteronismo remediable con glucocorticoides y la toxicidad por el regaliz.
Origen
Las células cromafínicas de las cuales los feocromocitomas surgen, son de origen neuroectodérmico y son parte del sistema nervioso adrenal (simpático adrenal). Son referidas como células cromafínicas debido al color oscuro que toman cuando sus grandes depósitos de catecolaminas son oxidadas en presencia de sales cromadas.
Tabla 1. Definiciones de las neoplasias de la médula adrenal.

Durante la sexta semana de vida, una porc\on del neuroectodermo adyacente al recientemente formado tubo neural, se separa y se sitúa en la región dorsal. Esta estructura es la cresta neura!, y sus células que subsecuentemente migran a lo largo del embrión desde la base del cráneo a la pelvis, efectúan una amplia variedad de funciones.
En el adulto, la mayoría de estas células están localizadas en la médula adrenal, aunque un grupo de ellas existe en los ganglios simpáticos, en el cuerpo carotídeo, en el glomus yugular. Son las mismas células C del tiroides secretoras de calcitonina, las de los islotes pancreáticos y las argentafínicas del intestino anterior (32). En personas jóvenes y en niños, una colección bien reconocida de estas células se localiza en posición paraórtica en nivel del origen de la arteria mesentérica inferior y forman lo que se conoce con el nombre de órgano de Zuckerkandl (12). Más tarde en la vida, estas células cromafínicas extraadrenales regresan, pero remanentes persistentes pueden ser el futuro locus para un feocromocitoma. Este origen explica porqué los feocromocitomas a veces están asociados con otros tumores o condiciones que surgen de fuentes neuroectodérmicas como el carcinoma medular del tiroides, la neurofibromatosis (la enfermedad de Von Recklinghausen), ganglioneuromatosis del tubo digestivo, neuroma de las mucosas, hemangioblastoma cerebeloso y angiomas retinales (enfermedad de Von Hippel-Lindau). Algunas de estas enfermedades embriológicamente relacionadas que surgen del mal desarrollo de la cresta neural. son referidas como neurocrestopatías e incluyen, además de las ya mencionadas, los neuroblastomas, tumores carcinoides, enfermedad de Hirchsprung, quimiodectomas y melanosis neurocutánea (5).
Los apudomas son tumores compuestos de células con aspectos citoquímicos y estructurales comunes. Encontrados en una variedad de tejidos endocrinos y neuroendocrinos que secretan hormonas polipéptidas o precursores hormonales; de aquí el acronímico APUD (amina, captación de ami nas precursores y descarboxilación).
Una clasificación de los tumores de la médula adrenal, su estado funcional y presentación clínica se muestra en la Tabla l. Como se puede analizar en la Tabla, el término feocromocitoma se usa para cualquier tumor funcional del sistema paraganglionar. Los tumores no funcionales de este sistema, se les denomina paragangliomas. Ambas categorías de neoplasias pueden ser adrenal o extraadrenal en origen, así como también benignas o malignas.
Biosintesis y secreción de catecolaminas
Las catecolaminas están ampliamente distribuidas en las plantas y en el reino animal. En los mamíferos, la adrenalina es sintetizada principalmente en la médula adrenal, mientras que la norepinefrina se encuentra no sólo en la médula adrenal, sino también en el sistema nervioso central y en los nervios periféricos simpáticos. La dopamina, precursora de la norepinefrina, existe en la médula adrenal y en neuronas noradrenérgicas. Está presente en altas concentraciones en el cerebro, en ganglios simpáticos con neuronas especializadas y en el cuerpo carotídeo, donde sirve como un neurotransmisor. También se encuentra en las células mastocitas especializadas y en las enterocromafínicas.
El sustrato inicial para la biosíntesis de las catecolaminas es la tirosina, la cual se puede derivar de los alimentos ingeridos o sintetizada en el hígado por hidroxilación a partir de la t”enilalanina (Diagrama 1).
La tirosina circula en una concentración de I a 1.5 mg/ 100 mL de sangre. Entra a las neuronas y a las células cromafínicas por un mecanismo de transporte activo, y es convertida en levodopa o dihidroxifenilalanina por la enzima tirosina hidroxilasa. Esta última enzima es importante porque de ella depende la cantidad de catecolaminas que se pueda producir. Sabiendo esta vía de biosíntesis se ha podido sintetizar un compuesto que funciona como un antagonista de la tirosina hidroxilasa y, por lo tanto, inhibe la producción de catecolaminas. La alfa metilparatirosina (metirosina) está ahora comercialmente disponible y ha probado ser útil en el tratamiento del feocromocitoma maligno. La levodopa es convertida a dopamina por la enzima L-aminoácido aromático descarboxilasa (dopa-de scarboxilasa). Esta sustancia se encuentra en todos los tejidos, pero con la más alta concentración en el hígado, riñones y cerebro. lnhibidores competitivos de la dopadescarboxilasa, tales como la alfa-metildopa, son convertidas por esta enzima en sustancias en forma de gránulos en las células nerviosas y liberadas, en lugar de la norepinefrina. Se pensó que estos productos (falsos neurotransmisores) mediaban la acción antihipertensiva de drogas en las sinapsis simpáticas periféricas, pero ahora se cree que estimulan los receptores alfa del sistema cortibulbar inhibitorio, reduciendo por lo tanto, las descargas simpáticas periféricas.
Diagrama 1. Biosíntesis y metabolismo de las catecolaminas a partir de la tirosina.
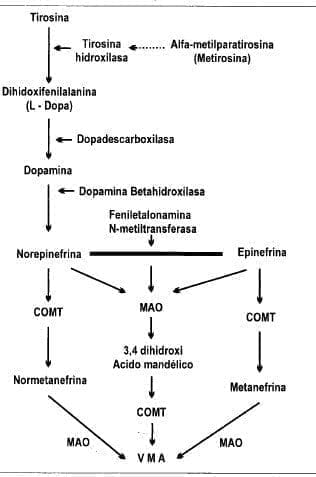 MAO+ Monoaminoxidasa; COMT = Catecol-o-metiltransferasa; VMA = ácido-vanilmendélico.
MAO+ Monoaminoxidasa; COMT = Catecol-o-metiltransferasa; VMA = ácido-vanilmendélico.
Tabla 2. Clasificación de los receptores adrenérgicos por selectividad de los agonistas y antagonistas.
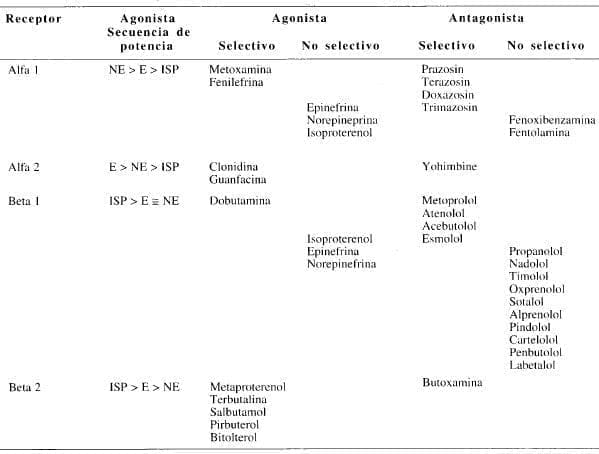 E = Epinefrina; NE = Norepinefrina; ISP = Isoproterenol.
E = Epinefrina; NE = Norepinefrina; ISP = Isoproterenol.
La conversión de dopamina a norepinefrina es catalizada por la dopamina beta-hidroxilasa. Esta enzima no se produce en tejidos fuera de las neuronas. La feniletalonamina- N-metil transferasa (PNMT) cataliza la metilación de la norepinefrina a epinefrina. Esta enzima se encuentra sólo en el citosol de las células de la médula adrenal y en muy pocas neuronas del sistema nervioso central. La PNMT es inducida por los altos niveles de glococorticoides (100 veces la concentración sistémica) encontrados en la médula adrenal como consecuencia de la organización vascular que perfunde las células medulares con sangre de la corteza
La iniciación de los efectos biológicos de las catecolaminas son extremadamente rápidos por su vida corta. Existen varios mecanismos que explican este hecho. Estos incluyen recaptación por las terminaciones nerviosas simpáticas, el metabolismo por las enzimas catecol-o-metil-transferasa y la monoaminooxidasa, la conjugación con sulfato y su excreción directa por el riñón (38). La catecol-o-metiltransferasa (COMT) transfiere un grupo metil a la epinefrina y norepinefrina, produciendo metanefrina y normetanefrina respectivamente. La monoaminoxidasa (MAO) cataliza la desaminación oxidativa de la epinefrina y norepinefrina a 3, 4 ácido dihidroximandélico. Este producto posteriormente es metabolizado por la COMT a ácido vanilmandélico (VMA). La metanefrina y noremetanefrina también son catalizadas por la MAO a VMA.
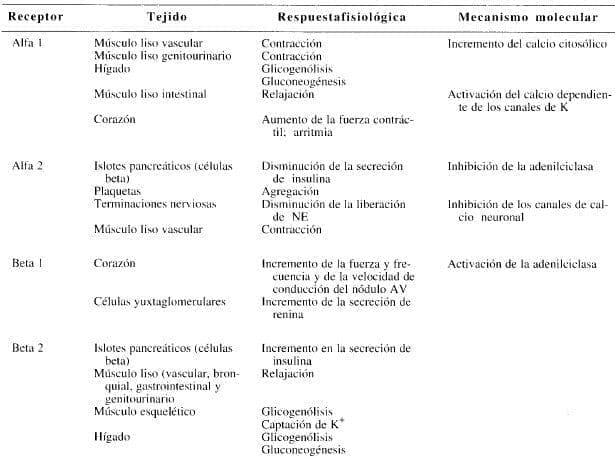
Tabla 3. Características de los subtipos de los receptores adrenérgicos.

me urge saber de un médico que opere los feocromacitoma. ..para un niño de 10 años por favor es urgente Gracias