Enfermedad Granulomatosa Crónica Ligada al Cromosoma X
Carlos Julio Montoya, Adriana Ayala
Grupo de Inmunodeficiencias Primarias
Facultad de Medicina, Universidad de Antioquia,
Medellín, Colombia.
Se presenta la historia clínica de un paciente que desarrolló desde los primeros meses de vida un síndrome de infección recurrente anormal severo. Con múltiples fístulas perianales de evolución crónica y varios episodios de sepsis por Salmonella spp.
Tres hermanos de sexo masculino habían fallecido en el primer año de vida por infecciones severas.
Los estudios de laboratorio realizados en el Laboratorio de Inmunología de la Universidad de Antioquia permitieron hacer el diagnóstico de la enfermedad y caracterizar el estado de portador de la madre.
Se desea resaltar la posibilidad de realizar diagnósticos específicos de inmunodeficiencias primarias en nuestro medio, partiendo de la detección de los pacientes con un síndrome de infección recurrente anormal.
Historia Clínica
Antecedentes personales
El paciente es producto de la cuarta gestación, de 9 meses de duración y con control prenatal sin alteraciones en la evolución. Parto vaginal institucional normal; peso, talla y examen físico posnatal normales. El cordón umbilical cayó en la segunda semana de vida. Recibió en los primeros meses las vacunas recomendadas en el esquema nacional, sin ninguna respuesta anormal.
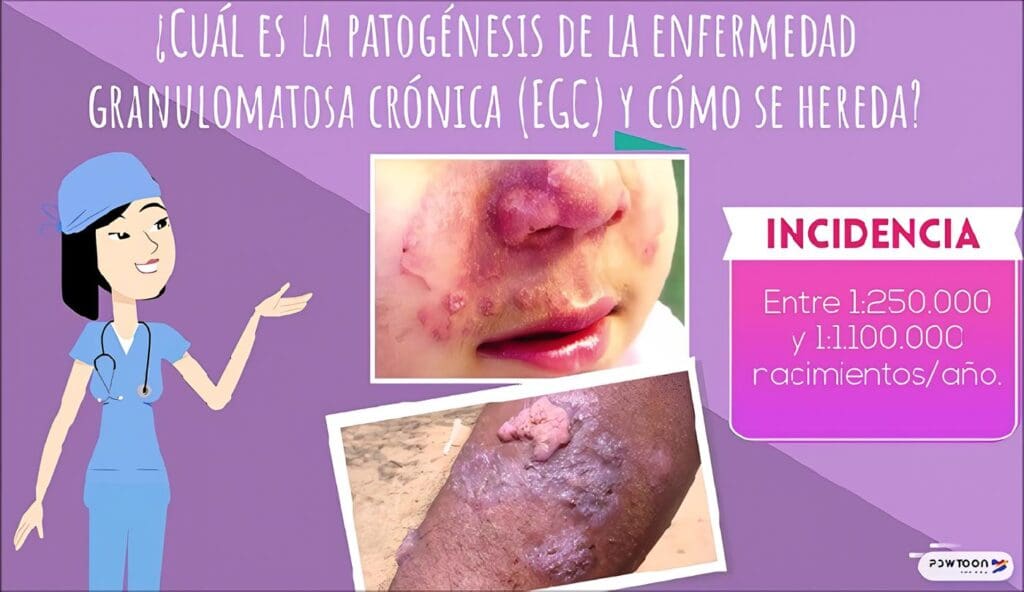
Desde el tercer mes de vida comienza la aparición de masas de carácter inflamatorio en la región perianal, que van aumentando de tamaño y finalmente supuran al exterior. Al parecer no se realizaron cultivos para determinar los gérmenes causantes.
Recibió tratamientos ambulatorios e intrahospitalarios (en la unidad de salud de su municipio) con Naproxén, Ampicilina, Penicilina G, Gentamicina y Metronidazol sin mejoría.
Durante varios episodios la fístulas perianales se aumentaron en tamaño y cantidad de supuración, presentando mejoría leve con el manejo antibiótico.
A los 6 meses de edad se encuentra, además de las fístulas, una hepatomegalia moderada y placas eritematosas descamativas en cuello. No presentaba en ese momento alte-raciones en el desarrollo pondoestatural.
En los siguientes meses es hospitalizado en múltiples ocasiones por recrude-cimiento de las fístulas. Recibiendo tratamiento antibiótico y sin estudios para caracterizar la etiología de las infecciones.
A los 17 meses de vida es remitido a la ciudad de Medellín para nueva hospitalización; realizan interconsulta al Programa Detección y Manejo del Síndrome de Infección Recurrente (Grupo de Inmunodeficiencias Primarias de la Universidad de Antioquia).
En la evaluación se encuentran múltiples fístulas perianales con escasa supuración; adenopatías supraclaviculares y axilares izquierdas de consistencia dura, móviles, no dolorosas, sin supuración; hepatoesplenomegalia no dolorosa. No tenía lesiones en piel.
Por los antecedentes familiares y el cuadro clínico se determinó descartar una inmunodeficiencia primaria con alteración en la función de las células fagocíticas
(Lea También: Evolución de la Enfermedad Granulomatosa Crónica)
Antecedentes familiares
En los años previos, tres hermanos de sexo masculino fallecieron durante el primer año de vida por cuadros de infecciones recurrentes severas. Fueron atendidos en una unidad hospitalaria local distante de Medellín, con manejo antibiótico y sin estudios de laboratorio para orientar el diagnóstico.
Todos presentaron predominantemente sintomatología del aparato digestivo (infecciones intestinales), y el último desa-rrolló fístulas perianales, sepsis y falla multiorgánica.
En éste se realizó a nivel local la autopsia. Pero careció de estudios microscópicos que revelaran la presencia de granulomas u otros hallazgos que orientaran al diagnóstico.
Hipótesis diagnóstica en la Enfermedad Granulomatosa Crónica
El desarrollo temprano de un síndrome de infección recurrente anormal del aparato digestivo, con desarrollo de fístulas perianales, hepato y esplenomegalia, y poliadenopatías sugirió una alteración en la inmunidad mediada por las células fagocíticas. En especial del tipo de una Enfermedad Granulomatosa Crónica (EGC), una Deficiencia en la Adhesión Leucocitaria. Un Síndrome de Chediak-Higashi, o una neutropenia congénita severa (Enfermedad de Kostmann).
Sin embargo, la falta de retardo en la caída del cordón umbilical y de úlceras crónicas necróticas no supurativas hacían sospechar más una EGC. La ausencia de fotofobia marcada y albinismo parcial iniciaban el proceso de descarte de un Síndrome de Chediak-Higashi
Estudios de laboratorio
Los hallazgos más relevantes de los múltiples estudios realizados al paciente son:
1. Estudio microscópico del cabello: ausencia de gránulos anormales
2. Diferentes hemoleucogramas realizados durante los períodos de infección mostraron unas cifras de leucocitos elevadas, con neutrofilia y bandemia.
Durante los episodios sin infección las cifras de leucocitos era normal. No se observaron gránulos anormales en los neutrófilos y otras líneas celulares. Una anemia moderada con microcitos se asociaba a las infecciones crónicas.
3. La evaluación inmunológica aportó los siguientes resultados:
-
- Cifras normales de linfocitos CD3+, CD4+, CD8+ y CD19+, determinados por citometría de flujo
- Expresión de las Beta 2 integrinas: CD11b y CD18 normal, determinada por citometría
- Electroforesis de proteínas normal
- Dosificación normal de inmunoglobulinas séricas
4. Otros estudios:
-
- Las pruebas de función renal y hepática fueron normales
- KOH y cultivo de lesiones cutáneas: Candida albicans
- Cultivo de material de las fístulas: polimicrobiano, sin predominio
5. Diagnóstico Fenotípico
-
- Reducción del NBT por neutrófilos en placa: 0% luego de la activación con PMA, lo que demostraba una ausencia en la activación del sistema NADPH oxidasa. La prueba se repitió en dos ocasiones para confirmar el diagnóstico de EGC. Esta prueba en la madre mostró un patrón en “mosaico”: el 51% de las células redujo el NBT, mientras el 49% no lo hizo. Esto confirmaba una EGC ligada al cromosoma X, con una madre portadora de la mutación.
- Evaluación de la explosión respiratoria de los fagocitos por citometría de flujo utilizando la Dihidrorodamina 123: ausencia de la producción de ROIs en el paciente (con relación a un control normal). En la madre, en esta prueba se observaron dos poblaciones de fagocitos: unos con producción de ROIS y otros sin explosión respiratoria.
Diagnóstico definitivo
Síndrome de Enfermedad Granulomatosa Crónica ligada al cromosoma X


Buenas noches tengo dos sobrinos mellizos con sospecha de EGC me gustaria saber que debemos hacer para poder realizar el test de dihidorodamina con ustedes
Buenas tardes Eliana, gracias por escribirnos.
Este es un contenido netamente informativo, nosotros somos un Portal de Contenido, por lo tanto no prestamos ningún tipo de servicio médico.
En el siguiente enlace puedes encontrar un directorio de Salud: https://encolombia.com/salud/dir-salud/
Saludos.