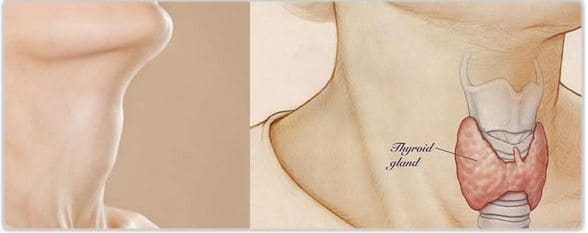
Fenotipo y hormonas sexuales
Desde hacía muchos siglos se habían informado casos ocasionales de personas cuyo aspecto causa confusión sobre su verdadero sexo.
Se dice que el poeta Ovidio –varias décadas antes de Cristo- habló de una adolescente griega que empezó a adoptar un fenotipo masculino, lo que posiblemente se trató de un síndrome adreno-genital. De aquella época, Séneca y Plinio el Joven describieron también casos curiosos de pubertad precoz.
El eunucoidismo era conocido de la antigüedad. Sin embargo, una descripción juiciosa de casos relacionados con un aspecto sexual inadecuado para la edad o el sexo sólo empezó a verse a partir del siglo XVIII y particularmente en el XX, cuando las nuevas tecnologías permitieron hacer mediciones hormonales en dichos casos.
Observaciones sobre estados intersexuales e hipogonadismo
A) Antes del siglo XX.
1753 Kästner (un matemático) describe una niña gorda de tres años, con pubertad precoz, que murió poco tiempo después. 1762 Morgagni describe un caso de ausencia congénita de ovario e infantilismo sexual. 1766 Von Haller informó el caso de una bebé que a los dos años menstruó y tuvo su embarazo a los ocho.
Recopiló dieciocho casos de la literatura, y abogó porque se dejara de lado la curiosidad de los casos, ya que un abordaje científico podría dar información sobre sus causas. 1803 Tiselius von Tilenau informó el caso de niña de cuatro años, pubertad precoz y un tumor suprarrenal a la autopsia. 1811. Cooke reportó un caso semejante al anterior. 1817 Steglehner describió el primer caso de un testículo feminizante.
A finales del siglo, en el conocido texto de Overzier, titulado La Intersexualidad –que trata de hermafroditismos verdades y seudohermafroditismos, Hauser incluye siete casos más de feminización de origen testicular.
1856 Aurelio Maestre de San Juan practicó una autopsia a un hombre hipogonádico de cuarenta años, con testículos muy pequeños y bulbos olfatorios ausentes. 1865 De Crecchio publicó el caso de un varón con hipospadias, genitales internos femeninos e hiperplasia suprarrenal. Ese mismo año se informó un caso de pubertad precoz en niña de tres años y un carcinoma suprarrenal.
1895 Eccole Sachi describió un niño de nueve años, completamente desarrollado, con un carcinoma testicular que al resecarse revirtió muchas de las manifestaciones de las características sexuales secundarias. 1896 Gutzeit describió pubertad precoz en un caso de teratoma de la pineal.
B) En la primera mitad del siglo XX
1904 Bouin y Ancel deducen el papel de las células de Leydig en la diferenciación fenotípica masculina. 1910 Apert publica una casuística de treinta y un enfermos con distrofias relacionadas con las cápsulas suprarrenales, en la que figuran algunos con hiperplasia suprarrenal. Se trata de personas con obesidad, amenorrea e hirsutismo.
Pelliizzi llama macrogenitosomia praecox a los casos –como el de Sachi- con pubertad precoz en los varones, pero más tarde se dice que si no se desarrollan las células germinales, se trata entonces de una pseudo-pubertad precoz. 1912 Gallais intenta clasificar los casos de pseudohermafroditismo.
1912 Kermauner considera que la ausencia congénita de ambas gonadas no deben llamarse varones sin testículos o mujeres sin ovarios, sino que se trata de una disgenesia de las gonadas. 1921 Achard y Thiers informan el caso de una mujer de setenta y dos años con barba, bigote, hipertensión, glucosuria, con manifestaciones de hipertricosis desde los diecinueve años y con hiperplasia suprarrenal bilateral a la autopsia.
1923 DeQuervain informó que ocasionalmente, al encontrar una gonada en sacos herniarios de niñas, se llevaba la sorpresa de que se trataba de testículos. 1930 Rossie describe un caso de ausencia congénita de ovario, que llama aplasia ovárica. Ese mismo año, Otto Ullrich informó un caso de anomalías fenotípicas similares a algunas descritas por él mismo por mutaciones en ratones, y considera que su paciente se parece a uno informado por Funke. Lo llamó trastorno de Bonnevie-Ullrich, que luego pasaría a ser mejor conocido como síndrome de Turner, disgenesia gonadal o monosomía X0.
El caso de Ullrich –vuelto a estudiar años más tarde- sí tenía cariotipo X0, así que no se trataba de un síndrome de Noonan, como algunos sugiereron.
1932 El neurocirujano Harvey Cushing publica un serie de pacientes con hiperfunción suprarrenal característica:
Lo que dio lugar a la enfermedad – y al síndrome- que llevan su nombre. Pocos años antes Weber y también Hunter, habían descrito sendos enfermos con variantes de los síndromes adrenogenitales, que incluían aspectos metabólicos como hiperglicemia, osteoporosis, hipertensión y también estrías.
1937 Albright describe la displasia fibrosa poliostótica, como una causa de pubertad precoz. 1938 Henry H. Turner –profesor de endocrinología en Oklahoma- publicó siete casos de talla baja, cuello alado, implantación baja del cabello, infantilismo sexual y cúbito valgo (Turner hizo mucho por la Endocrine Society, por la promoción de nuevas revistas de endocrinología y por el desarrollo de la investigación en la especialidad).
Catorce años después Albright –y también Barney- confirman el síndrome de Turner y observan aumento de las gonadotropinas en dichos pacientes, lo que indica la disgenesia ovárica; en 1959, el inglés Charles Ford encontró la anomalía cromosómica X0. En 1968, el pediatra J.A. Noonan describió un síndrome con fenotipo similar pero con cariotipo normal (XX o XY) y cardiopatía congénita, más frecuentemente estenosis mitral; un 50% de estos pacientes tienen una mutación del gen PTPN11.
Otros tres síndromes tienen hallazgos parecidos de malformación cardiaca y corta estatura postnatal con fenotipo de Turner: el LEOPARDO, el cardio-facio-cutáneo y el de Costello. 1939. Se publica en el Lancet el caso de la peruana Lina Medina, que tuvo su pubertad poco después de los tres años y un hijo a los cinco años, que nació por cesárea.
(Lea También: Hiperplasia Suprarrenal Congénita)
1943 Harry Klinefelter:
Describe nueve pacientes con hipogonadismo primario (ginecomastia, ausencia de espermatogénesis, preservación de células de Leydig y elevación en las gonadotropinas urinarias). Trece años más tarde Bradbury encuentra cromatina positiva en estos casos, por lo que se postula que tiene un cariotipo XXY.
Por estos años, el grupo de Fuller Albright en Boston –más conocido por sus estudios en el metabolismo óseo y la función de las paratiroides- se interesa también en los casos de Klinefelter, por lo que algunos llaman este síndrome como el de Klinefelter-Reifenstein-Albright.
Conrad Reifestein describe unos casos similares pero con cromosomas sexuales XY. También hay quienes llaman el síndrome de testículos feminizantes con síndrome de Reifestein. La sección de endocrinología del Massachussets General Hospital era muy activa en estudiar casos nuevos; la parte de tiroides la manejaba el famoso James Howard Means y la parte de metabolismo óseo, el recordado Fuller Albright.
Con el trabajó Reifestein y también Harry Ficht Klinefelter, quien venía de trabajar algo infructuosamente con Means. La primera vez que Klinefelter asistió a la consulta externa, se encontró con una paciente negro de diecinueve años, con ginecomastia y testículos pequeños. Albright lo encargó de que estudiara el caso, y luego los ocho más que aparecieron en la descripción inicial.
Klinefelter consideró que su síndrome debió haber llevado el nombre de su jefe Albright, quien generosamente le cedió el primer puesto en el artículo que contiene las primeras descripciones, a pesar de que este médico era el más joven e inexperto del grupo. Al terminar en Boston, trabajo por muchos años en el Johns Hopkins, donde se dedicó a la reumatología y a otros asuntos de la medicina, es decir, que no fue un endocrinólogo practicante.
1944 Novak considera que los casos de pubertad precoz en los que no se encuentra causa alguna deben denominarse constitucionales.
En realidad, estos son la mayoría de los casos. 1944 Franz Josef Kallmann –genetista germano-americano- describió tres familias con personas que tenían hipogonadismo y anosmia; estudios posteriores evidenciaron que se trataba de un hipogonadismo hipogonadotrófico, más claramente, de una deficiencia de la secreción de Gn-RH, asociada con atrofia del bulbo olfatorio. 1947 Del Castillo reconoce unos pacientes que –aunque tienen caracteres sexuales secundarios normales- tienen testículos pequeños, con azoospermia, túbulos seminíferos reducidos, ausencia completa del epitelio germinal y células de Sertoli normales.
Hermafroditismos y trastornos del cariotipo
A los pacientes que presentaban en forma simultánea características correspondientes a ambos sexos (estados intersexuales) se les empezó a denominar hermafroditas (recordando al hijo del dios Hermes y la diosa Afrodita). Se clasifican los casos en hermafroditismo verdadero (cuando se encuentra tanto tejidos ováricos como testiculares, algunas veces en la misma gónada, lo que se denomina ovotestis. Dicha condición es relativamente rara; El seudohermafroditismo, en cambio, es más común. Desde el punto de vista médico, el hermafroditismo sugiere dos factores:
Genitales Externos Ambiguos.
Son genitales que pueden no corresponder a la composición genética de la persona (por ejemplo: genitales femeninos en un paciente genéticamente masculino). Varias enfermedades pueden producir la aparición de genitales ambiguos y hermafroditismo. Puede haber anorquia o ausencia congénita de gonadas, gonadas disgenéticas como en el síndrome de Turner o en las disgenesis gonadales puras o mixtas, presencia de ambos tejidos –tanto testicular como ovárico- en el hermafroditismo verdadero.
Pseudohermafroditismo femenino (con gonada femenina normal) debido a masculinización por hormonas exógenas y otros mecanismos, durante la gestación (por ejemplo, progestágenos), o por hiperplasia suprarrenal congénita, en la que el 90% de los casos se debe a deficiencia de la 21-hidroxilasa. En el pseudohermafroditismo masculino, la gonada del varón es normal.
Entre estos casos se encuentran anomalías en la LH, algunos casos raros de hiperplasia suprarrenal congénita, los testículos feminizantes, los leydigomas -que en realidad causan una pubertad o macrogenitosomía precoz- y además hay otros trastornos del cariotipo como el síndrome de Klinefelter.