Fetos masculinos
Fetos masculinos
 Fetos masculinos
Fetos masculinosComo los fetos masculinos sufren una masculinización en presencia del andrógeno durante ciertos periodos prenatales se han estudiado sus efectos en casos clínicos específicos como la administración de progestágenos para abortadoras habituales o en las niñas con hiperplasia suprarrenal congénita.
Estas pacientes –particularmente las que tienen un síndrome adrenogenital- presentan masculinización parcial de sus genitales externos (como la hipertrofia clitoridiana) y de su conducta femenina; más frecuentemente son marimachas (lo que se conoce como tomboyismo) y prefieren los juegos de niños, tienen conductas poco maternales y escasas fantasías maternales, sienten menos satisfacción con la asignación de sexo femenino, son más robustas y despliegan más energía que niñas que sirvieron de controles; pero aunque no se ha encontrado una orientación homosexual, ni falta de deseo de casarse y tener hijos, posiblemente las mas masculinizadas –que sienten menor atracción heterosexual- tendrían más posibilidades de una eventual conducta homosexual o bisexual.
(Lea También: Hormonas y Homosexualidad)
Actualmente todos esos problemas tienden a omitirse, debido al diagnóstico precoz en las familias que llevan los genes alterados, y a tratamientos in utero.
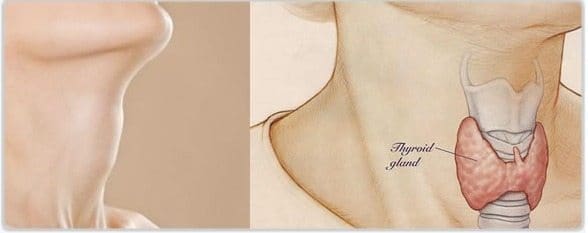
En 1950, tanto Lawson Wilkins como Bartter, descubrieron la posibilidad de suprimir el hiperandrogenismo de estos pacientes utilizando cortisona. Fue Bartter quien consideró que la hiperplasia, había un déficit en la producción de cortisol por algún bloqueo enzimático y una hiperproducción de ACTH que llevaba a la hiperplasia adrenal.
En 1953, Jailer planteó que se tratase de una deficiencia de la 21-hidroxilasa, en 1955 Walter Eberlein y Alfred Bongiovanni asignaron la etiología de los casos de virilismo con hipertensión a un a deficiencia de 11-hidroxilasa y en 1958, consideraron que los casos severos de deficiencia de la 21-hidroxilasa se manifestaban con insuficiencia suprarrenal severa y muerte probable, además de la virilización.
En 1961, Bongiovanni –del Hospital Pediátrico de Filadelfia- describió la deficiencia de 3-beta hidroxi-dehidrogenasa, con pseudo-hermafroditismo masculino en los hombres y trastornos electrolíticos severos.
Y en 1962, Frederick C. Bartter (y William Schwartz) comunicaron los datos correspondientes a dos pacientes con un síndrome caracterizado por hipertrofia e hiperplasia del aparato yuxta-glomerular, hiperaldosteronismo con presión arterial normal, alcalosis metabólica hipo-potasémica y defecto de la capacidad de concentración renal resistente a la acción de la pitresina.
En ambos sujetos se demostró un incremento de los niveles circulantes de angiotensina. La hiperplasia lipoídica fue descrita por Prader, y en 1966, Edward G. Biglieri describió los casos con deficiencia de 17-hidroxilasa.