La acidemia isovalérica es un ejemplo ya que con cambios menores, la mayoría de las enfermedades metabólicas se comportan con igual sintomatología y signología y su aproximación diagnóstica es la misma. Para el caso de esta acidemia, no se han descrito más de 80 casos en todo el mundo, desde la descripción original de la enfermedad por Tanaka en 1966, siendo además la primera enfermedad metabólica diagnosticada por medio de cromatografía de gases1.
Esquema Diagnóstico
El cuadro clínico de la enfermedad metabólica es polimorfo y comparte sintomatología variada con la de cuadros de hipoglicemia, neurológicos, de sepsis y desequilibrios ácido-básicos. Cuando no sea posible establecer con claridad el origen de estos cuadros, debe sospecharse la enfermedad de origen metabólico, y debe tomarse una muestra de suero y orina al ingreso, antes de iniciar cualquier tratamiento, que modifica las alteraciones bioquímicas, congelarlas y tenerlas disponibles para procesarlas posteriormente si el caso lo requiere.
La secuencia de exámenes de laboratorio que se sigue después de la sospecha clínica es: Glicemia, electrolitos, ácido láctico, amonio, función hepática y renal, y la RESERVA DE LAS MUESTRAS REFERIDAS.
Una ventana aniónica por encima de 15, hipoglicemia, acidemia láctica o hiperamonemia, con o sin función hepática alterada, son los primeros indicios para elaborar el diagnóstico. Si pasado el tiempo de estabilización del paciente, persiste la duda sobre la etiología, se procesarán, como pruebas de tamizaje, las muestras de orina y suero reservadas, para determinar en orina: Cloruro férrico, nitrosanaftol, dinitrofenilhidracina, nitroprusiato y azúcares reductores, que en forma general y cualitativa indican presencia de aminoácidos y/o sus metabolitos y la presencia de azúcares con la última. Y en sangre y orina la determinación de aminoácidos y de ácidos orgánicos por cromatografía en capa fina. Ya es posible procesar estos exámenes en varios laboratorios de las principales capitales del país. Haciendo el análisis de este caso clínico, se vislumbrarán las características para la sospecha y el diagnóstico de las enfermedades metabólicas en general.
Aproximadamente la mitad de los casos reportados de acidemia isovalérica tienen una presentación aguda en el período neonatal, con una alta mortalidad. La otra forma de presentación es crónica intermitente, correspondiendo esta variedad al caso que se presenta. El niño desencadenó la crisis posterior a una comida rica en proteínas, durante la reunión familiar mencionada anteriormente y expresó a continuación la sintomatología típica descrita para la enfermedad: Vómito, letargia que progresa a coma, acidosis metabólica y cetonuria, que al ingreso fueron interpretados como parte de un cuadro infeccioso o metabólico (sepsis, síndrome de Reye o acetoacidosis diabética); y neurológico cuando fue remitido con diagnóstico de una lesión traumática del SNC. Estos hallazgos clínicos son inespecíficos y se pueden presentar en prácticamente todas las enfermedades metabólicas. El olor particular es uno de los pilares del diagnóstico y es descrito como un olor a “sudor de pies” debido a las concentraciones altas de ácido isovalérico; fué percibido al ingreso por el personal de enfermería, pero solo se valoró retrospectivamente. Persistió varios días, siendo más notorio en las mañanas. A pesar que es importante para llegar al diagnóstico, el olor no es exclusivo de esta acidemia y se puede encontrar también en la aciduria glutárica Tipo II1. Así mismo otras enfermedades también tienen olores característicos correspondientes a la acumulación de los metabolitos acumulados.
La mayoría de enfermos presentan neutropenia, trombocitopenia y pancitopenia, estos hallazgos ayudaron al diagnóstico erróneo de sepsis de origen no claro. La causa de las citopenias es probablemente la acción del ácido isovalérico sobre células progenitoras a nivel de la médula ósea1. Esta pancitopenia favorece la aparición de infecciones, pero tiende a ser transitoria, corrigiendo al compensarse la crisis6. En algunas oportunidades puede simular una leucemia promielocítica, secundaria al efecto de los metabolitos anormales sobre células progenitoras de granulocitos7.
La enfermedad debe considerarse en cualquier neonato o niño mayor que tenga vómito, hiporexia, letargia, coma, acidosis metabólica, cetosis, hiperamonemia, hipocalcemia, neutropenia y trombocitopenia. El diagnóstico de certeza se establece con el análisis de ácidos orgánicos en plasma y en orina, preferiblemente en ésta última. Se busca la presencia de altas cantidades de isovalerilglicina, con una menor elevación del ácido 3-hidroxivalérico 1,8. Se puede determinar la actividad de la enzima por métodos fluorométricos en fibroblastos1. Es factible hacer el diagnóstico prenatal, bien sea cuantificando la actividad de la enzima en amniocitos o midiendo los niveles de isovalerilglicina en líquido amniótico8.
El tratamiento incluye además de las medidas generales, la infusión de glucosa como fuente principal de calorías y en algunos casos de bicarbonato para el control de la acidosis. Se debe restringir la ingestión de proteínas en la dieta e idealmente dar un suplemento libre o bajo en leucina6,9. Una de las partes principales del tratamiento consiste en la administración de suplementos de carnitina y glicina, que tienen como fin convertir el tóxico isovaleril-CoA, en los compuestos no tóxicos y fácilmente excretables por orina, isovalerilglicina e isovalerilcarnitina2,6,9. En este caso sólo fue posible administrar suplemento de carnitina, ya que la glicina no se consigue en nuestro medio.
El objetivo principal del manejo nutricional es reducir los aportes de Leucina para disminuir los niveles sanguíneos1. Por ser la leucina un a.a. esencial es necesario aportarlo en las cantidades mínimas suficientes para lograr un crecimiento adecuado y evitar la toxicidad. Se estima que los requerimientos mínimos de Leucina son de 100 mg/k/día en lactantes hasta 3 meses y de 40 a 50 mg/k/día al año de edad1.
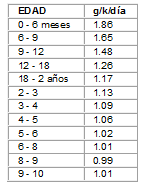
Una restricción de proteínas (lactantes 2 g/k/día y niños mayores 1 a 1.5 g/k/día), combinada con ingesta adecuada de calorías, carnitina y glicina, son suficientes para disminuir la producción de ácido isovalérico. Sin embargo, estas dietas restringidas, deben aportar las cantidades mínimas de nitrógeno y a.a. esenciales para cubrir las necesidades para el normal crecimiento y desarrollo (Tabla No. 1).
Tabla No. 1 Ingesta mínima de proteínas para lactantes y niños. Nivel de seguridad *
- Proteínas de alto valor biológico, (leche y huevo) 100% de digestibilidad.
Es necesario proveer una adecuada ingesta energética para garantizar adecuada síntesis protéica y evitar el catabolismo protéico y la elevación de leucina, mediante el consumo de alimentos elaborados con bajo contenido de este a.a. y suplementos energéticos. Se deben evitar los períodos largos de ayuno y en estos casos se deben garantizar los aportes con alimentación enteral con sonda.
La suplementación de micronutrientes es indispensable. Hierro, cobre, zinc, calcio y vitaminas del complejo B, así como de carnitina y glicina. Durante los episodios agudos interrecurrentes, la dieta debe estar exenta de proteínas y cubrir las necesidades de calorías con hidratos de carbono y grasas.
Las crisis tienden a disminuir con el paso de los años, posiblemente por la menor cantidad de infecciones, y por una ingesta proporcionalmente menor de proteínas6. Aunque pueden tener un desarrollo psicomotor normal, la mayor parte de los enfermos presentan algún grado de retardo psicomotor o congnitivo2. El paciente reportado presenta un retardo psicomotor leve y un retardo del desarrollo del lenguaje.
Es importante resaltar, que la sospecha de un error innato del metabolismo, se fundamentó en este caso, no sólo en la sintomatología del ingreso, también en el antecedente de consanguinidad de los padres, de un episodio similar meses antes y en el hecho de que el cuadro se inició luego de una comida abundante en proteínas, características estas, que son afines a una gran cantidad de defectos del metabolismo de los aminoácidos10. Dependiendo de la mutación, el defecto enzimático correspondiente es variable y esto determina que las enfermedades se presenten en una forma distinta en el período de recién nacido o que algunas veces los síntomas pasen desapercibidos y se manifiesten más tarde.
Bibliografía
1. Sweetman L, Williams J. Branched chain organic acidurias. En Scriver C, Beaudet A, Sly W, Valle D (eds): The metabolic and molecular bases of inherited disease, Vol II. New York, McGrawHill, 1995;(II):1393-7.
2. Berry G, Yudkoff M, Segal S. Isovaleric adicemia: Medical and neurodevelopmental effects of long-term theraphy. J Pediatr 1988;113:58-64
3. Lemieux B. Auray Blays C. Giguere R. Shapcott D. Newborn urine screening experience with over one million infants in the Quebec Network of Genetics Medicine. J. Inher. Metab. Dis. 1988;11:45-55.
4. Thomas G. Howell R. Selected screening Test for Genetic Metabolic Diseases. Year Book Medical Publishers. Inc. Chicago. 1973.
5. Chalmers RA. Lawson AM. Organic Acids in man. Analistical Chemistry biochemistry and diagnosis.of the organic acidurias. 1982; Chapmann and Hall. London.
6. Mayatepek E, Kurcznski T. Hoppel C. Long-term L-carnitine treatmen in isovaleric acidemia. Pediatr Neurol 1991; 7: 137-40.
7. Gilbertt-Barness E. Barness LA. Isovaleric acidemia with Promielocytic Myeloproliferative Synddrome. Pediat. Develop. Pathol. 1999; 2: 286-91.
8. Shigematsu Y, Hata Y, Nakai A, Kikawa Y, Sudo M, Tanaka Y, et al Prenatal diagjnosis of organic acidemias bases on amniotic fluid levels of acylcarnitines. Pediatr Res 1996; 39: 680-4.
9. Fries M, Rinaldo P, Schmidt-Sommerfeld E, Jurecki E, Packmn S. Isovaleric acidemia: Response to a leucine load after weeks of supplementation with glycine, L-carnitine, and combined glycine-carnitine therapy. J Pediatr 1996; 129: 449-52.
10. Mehta K, Zsolway K, Osterhoudt K, Krantz Y, Henretig F, Kaplan P. Lessons from the late diagnosis of isovaleric acidemia in a five-year-old boy. J Pediatr 1996; 129: 309-10.