Paciente de sexo femenino, 75 años, quien consulta por presentar cuadro de 15 días de evolución consistente en tos seca y posterior dolor dorsal, fiebre, tos seca emetizante y anorexia. Refiere sudoración profusa los últimos 3 meses. Antecedente de cuadro clínico y radiológico similar 2 años antes, manejada hospitalariamente como neumonía multilobar con cefuroxima mejorando clínica y radiológicamente.
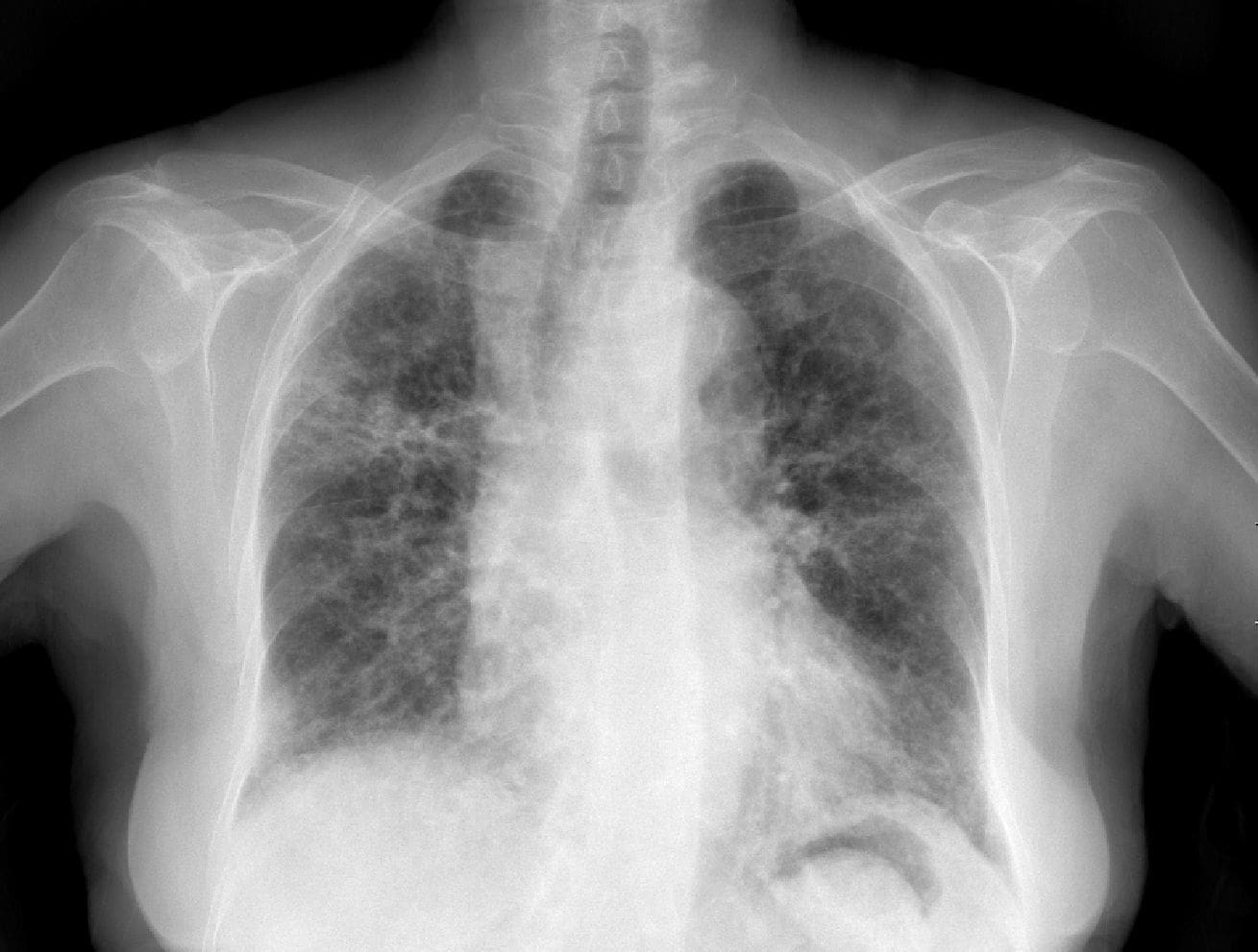
El examen clínico demostró una paciente en regular estado, con tos seca frecuente, sin adenopatías. No había alteraciones a la auscultación cardíaca o pulmonar, ni Hipocratismo digital. No había edemas. El Cuadro Hemático no mostró leucocitosis y el recuento blanco fue normal. Los Rayos X de tórax demostraron infiltrados bilaterales a parches, que comprometían especialmente el lóbulo medio, la base derecha, y los lóbulos inferiores.
Se manejó como neumonía multilobar con Ceftriaxona y Claritromicina con mejoría de la fiebre y la anorexia, pero persistencia de la tos seca. En vista de la distribución atípica de los infiltrados, y del antecedente de episodio similar se solicitó TAC de tórax de alta resolución que muestra infiltrados densos irregulares a parches, principalmente periféricos (Figura 2). Se decidió practicar biopsia pulmonar a cielo abierto. (Lea también: Fisiopatología del tromboembolismo pulmonar)
 Figura 2. TACAR de tórax que demuestra compromiso bilateral parchoso de predominio periférico
Figura 2. TACAR de tórax que demuestra compromiso bilateral parchoso de predominio periférico
*Dr. Horacio Giraldo Estrada, **Dra. Paulina Ojeda León
* Médico Internista Neumólogo, Profesor asistente Universidad El Bosque
** Jefe Depto. Patología Hospital Santa Clara. Profesora Universidad El Bosque
Evolución
La biopsia abierta de pulmón mostró engrosamiento septal difuso por hipercelularidad a base de linfocitos e histiocitos, con leve fibrosis focal, los alvéolos se encontraron revestidos de neumocitos hiperplásicos con luz libre, diagnosticándose un patrón de Neumonía Intersticial No Específica tipo II, con componente inflamatorio y fibrosis.
Discusión
Las Neumonitis Intersticiales Idiopáticas (NII) comprenden un número de entidades clínico-patológicas suficientemente diferentes entre sí, por lo que deben designarse como entidades separadas. El enfoque diagnóstico debe incluir una evaluación clínica, radiológica y patológica (1).
La evaluación clínica debe incluir, además de una historia detallada de los síntomas y el tiempo de evolución, una búsqueda de patologías coexistentes tales como enfermedades del colágeno o infección por el Virus de Inmunodeficiencia Humana (VIH).
Debe incluirse un interrogatorio completo sobre todo tipo de exposición incluyendo tabaquismo y uso de drogas o tóxicos, así como exposiciones en el ambiente laboral. Al examen clínico debe explorarse la presencia de estertores y de hipocratismo digital. La presencia de inflamación articular o engrosamiento de la piel puede orientar hacia una enfermedad del colágeno.
TACAR en neumonitis intersticial
El estudio debe continuar con una Radiografía de tórax y Pruebas funcionales pulmonares. Si estos estudios orientan hacia un diagnóstico de enfermedad intersticial, se debe practicar una Tomografía Axial Computarizada de Alta Resolución (TACAR) del tórax.
Si el patrón clínico y radiológico (TACAR) sugiere en forma clara la presencia de Fibrosis Pulmonar Idiopática / Neumonitis Intersticial Usual, lo cual requiere usualmente un Clínico y Radiólogo con experiencia en este tipo de pacientes, puede evitarse en forma razonable la biopsia pulmonar abierta.
La TACAR típica de la Neumonitis Intersticial Usual muestra un patrón reticular bilateral, predominantemente basal y subpleural, asociado con quistes subpleurales (patrón de panal de abejas) o bronquiectasias por tracción, y ausencia de consolidaciones y de nódulos, lesiones que van desapareciendo hacia los ápices.
Cuando el diagnóstico radiológico se basa en estos hallazgos, la precisión es superior al 90% de los casos. Cuando los hallazgos a la TACAR sugieren la presencia de enfermedades como Sarcoidosis, Neumonitis por hipersensibilidad, Linfangioleiomiomatosis, Histiocitosis de Células de Langerhans o Proteinosis Alveolar Pulmonar, el procedimiento de elección es una Broncoscopia con Lavado Broncoalveolar y Biopsia Transbronquial.
Por el contrario, cuando los hallazgos son “atípicos” (compromiso de los lóbulos superiores o peribroncovascular, patrón predominante de vidrio esmerilado o presencia de micronódulos), o cuando hay datos clínicos “atípicos” (pacientes jóvenes, ausencia de historia de exposición, ausencia de disnea o de defecto restrictivo en las pruebas de función pulmonar, o hay severa linfocitosis al LBA), está indicada la biopsia pulmonar.
Clasificación actual histológica
La clasificación histológica actual se divide en 7 grupos (1):
1.Fibrosis Pulmonar Idiopática / Alveolitis Fibrosante Criptogénica (FPI/AFC)
2. Neumonitis Intersticial No Específica (NINE)
3. Neumonitis en Organización Criptogénica (NOC)
4. Neumonitis Intersticial Aguda (NIA)
5. Enfermedad Pulmonar Intersticial con Bronquiolitis Respiratoria (EPIBR)
6. Neumonitis Intersticial Descamativa (NID)
7.Neumonitis Intersticial Linfoide (NIL)
Neumonitis Intersticial No Específica
El término neumonitis intersticial no específica se utilizó hacia 1980 para la Neumonitis no infecciosa presente en los pacientes infectados con VIH.
Posteriormente en 1994 Katzenstein y Fiorelli (2) utilizaron el término para designar algunos casos de enfermedad intersticial que no cabían dentro de los grupos ya bien definidos de patología histológica intersticial, y ha servido para definir un grupo de pacientes con enfermedad diferente a la Fibrosis Pulmonar Idiopática, y de un poco mejor pronóstico que esta, y que deben diferenciarse también de la NID, NIA, y la NOC3.
Katzenstein y Fiorelli dividieron la NINE en tres subgrupos dependiendo de la cantidad de inflamación o fibrosis presente en las biopsias pulmonares: Grupo I: con inflamación intersticial, Grupo II: con inflamación y fibrosis, y Grupo III: con fibrosis (3).
Clínicamente, no se ha definido completamente un cuadro típico de esta entidad. La edad promedio de inicio es entre los 40 50 años, con curso clínico lentamente progresivo e intermitente. No tiene predominancia de sexo, ni se ha asociado al consumo de cigarrillo.
El promedio de duración de síntomas antes del diagnóstico son entre 18 y 31 meses, con evoluciones tan cortas como 6 meses o tan largas como 3 años. Los síntomas usuales son tos, disnea y fatigabilidad y hay pérdida importante de peso en casi la mitad de los casos.
Síntomas constitucionales
Síntomas constitucionales como fiebre son ocasionales, y el hipocratismo digital se presenta en el 10 a 35% de los casos. Pueden escucharse estertores basales o difusos. Puede presentarse como única manifestación clínica meses o años antes de que se desarrolle una enfermedad del colágeno. La mortalidad estimada es de 15 a 20% en 5 años.
Funcionalmente, se encuentra un trastorno ventilatorio restrictivo usualmente menos severo que el encontrado en pacientes con FPI, con disminución de la Capacidad Pulmonar total (CPT) y del Volumen Residual (VR).
La Difusión al Monóxido de Carbono (DLCO) está disminuida en todos los casos, y en la mayoría se desarrolla hipoxemia con el ejercicio. El Lavado Bronco Alveolar (LBA) demuestra aumento de los linfocitos en 50% de los casos, y puede haber incremento similar en los polimorfonucleares y los eosinófilos.
Radiológicamente se encuentra compromiso bilateral, de predominio hacia las bases. Las opacidades parenquimatosas parchosas son el patrón más frecuente (4), pero puede presentarse también como patrón intersticial.
TACAR
En la TACAR el patrón de vidrio esmerilado es el más frecuente, con distribución bilateral y simétrica, de predominio subpleural. Pueden verse opacidades lineales o reticulares y asociarse a bronquiectasias por tracción. La presencia de panal de abejas o consolidación pulmonar es rara. En la mayoría de los casos que se ha podido hacer seguimiento estas anormalidades mejoran.
Histológicamente la NINE comprende una amplia gama de alteraciones parenquimatosas con diferentes grados de inflamación de la pared alveolar y fibrosis, predominando cualquiera de estos dos patrones, o en una combinación de ambas.
El intersticio alrededor de los vasos, bronquios y pleura puede estar comprometido. La inflamación usualmente es de tipo crónico, dependiente de linfocitos con escasas células plasmáticas. Fibrosis organizativa en la luz de los alvéolos puede verse en 2/3 partes de los casos pero es menor que la presente en el patrón de Neumonía organizativa.
El tratamiento debe iniciarse tan pronto se haga el diagnóstico, con el fin de evitar el desarrollo de fibrosis irreversible, y debe prolongarse indefinidamente en los casos en que el paciente mejora o está estable.
Debe revalorarse completamente el caso cada 6 meses, y de demostrar empeoramiento, debe considerarse otro medicamento citotóxico o llevarlo a trasplante pulmonar.
Tratamiento con corticosteroides
El tratamiento se hace con corticosteroides asociados con Azathioprina o Ciclofosfamida a las siguientes dosis (5):
Corticoides (Prednisona): 0.5 mg/Kg (peso ideal)/día por 4 semanas, seguidos de 0.25 mg/Kg (peso ideal)/día por 8 semanas, seguidos de 0.25 mg/Kg (peso ideal)/día por medio.
Azathioprina: 2-3 mg/Kg (peso ideal)/día, máximo 150 mg/día por vía oral. Debe iniciarse 25 a 50 mg/día e incrementar 25 mg día cada 1 a 2 semanas hasta llegar a la dosis máxima.
Ciclofosfamida: 2 mg/Kg (peso ideal)/día, máximo 150 mg/día por vía oral. Debe iniciarse 25 a 50 mg/día e incrementar 25 mg día cada 1 a 2 semanas hasta llegar a la dosis máxima.
Bibliografía
1.American Thoracic Society / European Respiratory Society. International Multidisciplinary Consensus. Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2002; 165:277-304
2.Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994; 18:136-147.
3.Nagai S, Kitaichi M, Itoh H, Nishimura K, Izumi T, Colby TV. Idiopathic nonspecific interstitial pneumonia/fibrosis: comparison with idiopathic pulmonary fibrosis and BOOP (corrigendum: Eur Respir J 1999; 13:171). Eur Respir J 1998; 12:1010-1019.
4.Park JS, Lee KS, Kim JS, Park CS, Suh YL, Choi DL, Kim KJ. Nonspecific interstitial pneumonia with fibrosis: radiographic and CT findings in seven patients. Radiology 1995; 195:645-648.
5.American Thoracic Societv. Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment. International Consensus Statement. Am J Respir Crit Care Med 2000; 161:646-664