A propósito de 2 casos clínicos
GUSTAVO GÓMEZ TABARES*, CAROLINA RENDÓN RESTREPO**
Resumen
Amenorrea primaria en una mujer con características sexuales secundarias presencia de ovarios y cariotipo 46XX son descritos en dos casos, uno con ausencia de formación de útero y otro con útero cavitado y antecedentes de consulta por dismenorrea severa cíclica con detección de hematómetra, ambos con agenesia de vagina.
Se hace revisión del síndrome y se describen sucintamente las técnicas quirúrgicas reconstructivas de vagina y de neocuello con bibliografía donde se conocen las técnicas quirúrgicas detalladas.
Summary
Primary amenorrhea in a woman with presence of secondary sex characteristics and ovaries and karyotype 46XX, is described in two cases, one with no formation of the uterus and one with cavitated uterus and background of cyclic severe dysmenorrhea and findings of hematómetra, both with vaginal agenesis.
It makes a revision of the syndrome and vaginal and neocérvix reconstructive surgical technic with bibliography where surgery techniques are described
Introducción
La agenesia mulleriana ocurre en una de cada 4.000 a 10.000 mujeres. La presentación más común es la ausencia congénita de vagina, útero o ambos, lo cual se denomina aplasia mulleriana, síndrome de Mayer Rokitansky Kuster Hauser (MRKH) o agenesia vaginal1.
El síndrome fue descrito por primera vez por Mayer (1829), y luego por Rokitansky (1838), como la agenesia del útero y la vagina debido a un desarrollo anormal de los conductos mullerianos; más adelante Kuster (1910) informó que esta entidad se asociaba frecuentemente con malformaciones urológicas, de ahí su nombre MRKH2.
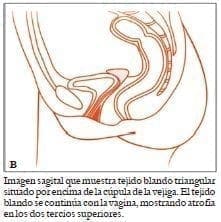
El síndrome de MRKH se caracteriza por aplasia congénita del útero y los dos tercios superiores de la vagina, en mujeres con desarrollo normal de las características sexuales secundarias y un cariotipo normal (46 XX). MRKH puede presentarse solo tipo I, o en mayor medida asociado a defectos renales, vertebrales, y en algunas ocasiones con defectos auditivos y cardiacos tipo ii o asociación MURCS.
El primer signo del síndrome de MRKH es una amenorrea primaria en una mujer joven, con desarrollo normal de las características sexuales secundarias, genitales externos normales, ovarios normales y funcionales, con cariotipo 46 XX, sin una anomalía cromosómica visible.
Sus manifestaciones fenotípicas se superponen con otros síndromes y asociaciones, por lo que requiere una caracterización precisa. Por mucho tiempo ha sido considerada como una anomalía esporádica, pero el incremento en el número de casos familiares ahora apoyanj una causa genética.
En casos familiares, el síndrome parece ser transmitido como un rasgo autosómico dominante, conpenetrancia incompleta y expresividad variable. Sin embargo, la etiología del síndrome de MRKH aún no está clara. El tratamiento de la aplasia de vagina consiste en la creación de una neovagina, que se realiza para permitir el coito.
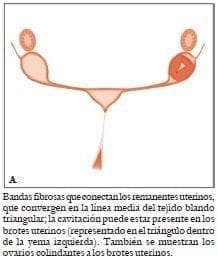
Debido al impacto psicológico del síndrome de MRKH en las mujeres jóvenes, es importante asesorar a las familias antes y después del tratamiento14. Dibujos esquemáticos de los típicos remanentes de conductos de Müller13
Nombre de la enfermedad y sinónimos
El síndrome de MRKH ha sido nombrado como CAUV (ausencia congénita de útero y vagina), MA (aplasia mulleriana) o GRES (síndrome genital-renal-auditivo) 14.
Definición y criterios diagnósticos
El síndrome de MRKH se caracteriza por aplasia congénita del útero y los dos tercios superiores de la vagina, en mujeres con desarrollo normal de las características sexuales secundarias y un cariotipo normal 46 XX14.
Otras malformaciones asociadas incluyen (tipo II o asociación MURCS):
- Malformaciones renales: agenesia unilateral, ectopia, riñones en herradura.
- Malformaciones esqueléticas: en especial vertebrales (anomalía de Klippel Feil; vértebras fusionadas, principalmente cervicales; escoliosis).
- Defectos de audición.
- Malformaciones cardiacas (menos frecuentes).
- Anomalías en los dedos, sindactilias, polidactilias (menos frecuentes).
Epidemiología
La incidencia es de una por cada 4.500 mujeres. La mayoría de los casos parecen ser esporádicos, aunque también se han descrito casos familiares. La herencia es autosómica dominante, con penetrancia incompleta y expresividad variable; lo cual sugiere que la prevalencia del síndrome puede ser subestimada. El tipo I es menos frecuente que la asociación MURCS14. (Ver también: Endocrinología Ginecológica)
Clínica
Generalmente se manifiesta con amenorrea primaria en adolescentes con crecimiento y desarrollo normales1. Estas pacientes tienen un fenotipo femenino, un cariotipo normal (46 XX), ovarios normales y funcionales, sin signos de exceso de andrógenos. El examen físico evidencia pubertad completa, con características sexuales secundarias (desarrollo del vello púbico y mamas TANNER V) y genitales externos normales. Simultáneamente la vagina tiene una profundidad menor de 2-7 cm.
El 2-7% de las pacientes con agenesia mulleriana tienen endometrio activo en las estructuras uterinas; estas pacientes presentarán dolor pélvico crónico o cíclico1.
El examen físico es necesario para diagnosticar el síndrome de MRKH, en cualquiera de sus dos tipos. La aplasia uterina en presencia de dos cuernos rudimentarios rodeados del repliegue peritoneal y trompas de Falopio normales corresponde al síndrome MRKH tipo I. El tipo II se caracteriza por hipoplasia uterina simétrica o asimétrica, acompañada de aplasia de uno o ambos cuernos, o diferencia en el tamaño de los dos cuernos rudimentarios, y malformaciones tubáricas como hipoplasia o aplasia de una o ambas tubas14.
También se han descrito casos con ovarios poliquísticos y tumores ováricos. Por otra parte, se ha descrito aplasia o ausencia de conductos mullerianos sugestivos del síndrome de MRKH, en casos de disgenesia o agenesias gonadales, en pacientes con fenotipo femenino con XY o XO. Hasta el momento, estas alteraciones gonadales no se consideran parte del síndrome de MRKH, mostrando una asociación aleatoria14.
* Ginecólogo y endocrinólogo. Profesor titular y distinguido de la Universidad del Valle. Director de la Clínica Endocrinología Ginecológica y Menopausia, Hospital Universitario del Valle, Cali, Colombia.
** Residente de Ginecología y Obstetricia. Rotante por Clínica Endocrinología Ginecológica HUV. Universidad Surcolombiana, Neiva, Colombia.