Jonathan A. Sánchez1, Susana Gómez2, Cristian Morales1, Sergio Iván Hoyos3
Palabras clave: conducto colédoco; quiste del colédoco; ampolla hepatopancreática; neoplasias del sistema biliar; anastomosis quirúrgica.
Resumen
Los quistes del colédoco son una condición médica poco frecuente, la cual se caracteriza por presentar dilatación de la vía biliar intrahepática y extrahepática; se postulan en su etiología la anomalía de la unión pancreático-biliar, lo que favorece el reflujo del jugo pancreático al árbol biliar, y la aganglionosis del árbol biliar. Tiene una amplia gama de presentación de síntomas, y entre los principales se encuentran el dolor abdominal, la ictericia y una masa abdominal palpable. Son lesiones premalignas y, por ende, el tratamiento de elección es la resección quirúrgica completa con seguimiento a largo plazo.
Introducción
Los quistes del colédoco son dilataciones quísticas de la vía biliar extrahepática, la intrahepática o ambas, con repercusión en la función hepática a corto o largo plazo. Fueron descritos en 1723 por Vater y Ezler, y la primera resección quirúrgica de un quiste del colédoco fue realizada en 1924 por MacWhorter 1. En Europa y en Estados Unidos, es una enfermedad poco frecuente; la mayor prevalencia se presenta en Asia oriental, especialmente en Japón 2. La tríada clásica de ictericia, masa abdominal palpable y dolor abdominal, debe hacer sospechar el diagnóstico en los niños. En los adultos, se relaciona más frecuentemente con síntomas comunes de enfermedad hepático-biliar, como son ictericia, colangitis, litiasis, de neoplasia coledociana y de pancreatitis 3,4. Los síntomas se derivan de la alteración estructural del conducto biliar 5,6.
Epidemiología
Los quistes del colédoco son una condición médica rara, con una incidencia en la población occidental de 1 en 100.000 a 150.000 nacidos vivos por año, aunque se ha informado que puede ser tan alta como 1 de cada 13.500 nacidos en los Estados Unidos y 1 de cada 15.000 nacimientos en Australia 6. Por razones desconocidas, la incidencia es mayor en poblaciones asiáticas, donde las cifras son tan altas como 1 en 1.000 nacidos vivos, 2 siendo Japón donde ocurren cerca de dos tercios de todos los casos 2. Tienen un claro predominio en las mujeres, con una relación de 4:1 7. La edad media de presentación es entre los 20 y los 30 años 7. Los síntomas predominantes de presentación en el adulto son el dolor abdominal y la ictericia 7, aunque también se puede manifestar como masa abdominal y, en el 15 %, con la tríada clásica de dolor abdominal, masa e ictericia. La incidencia de colangitis es de 15 % y en 26 % la presentación inicial es la coledocolitiasis 8.
Clasificación
Alonso-Lej propuso el primer sistema de clasificación para los quistes del colédoco, en 1959 9, con tres tipos de dilatación del conducto biliar. En 1977, Todani amplió este sistema e incluyó los quistes intrahepáticos y los múltiples 10. Actualmente, esta clasificación es la más aceptada y comprende cinco tipos de quistes, según su morfología y localización (figura 1).
 Figura 1. Clasificación de Todani
Figura 1. Clasificación de Todani
El tipo I consiste en la dilatación fusiforme de la vía biliar extrahepática. Los quistes de tipo I se subclasificaron posteriormente en tres tipos. En el tipo IA existe una importante dilatación quística extrahepática de todo el árbol biliar con preservación de los conductos intrahepáticos, y el conducto cístico y la vesícula biliar surgen de la dilatación del conducto biliar común; en el tipo IB hay dilatación focal y segmentaria de la vía biliar extrahepática por debajo de la unión hepático-cística; y, finalmente, en el tipo IC existen dilataciones lisas y fusiformes de todo el conducto biliar extrahepático, que usualmente se extienden desde la unión pancreático-biliar hasta el árbol biliar intrahepático. El tipo II corresponde al divertículo extrahepático supraduodenal. El tipo III es el denominado coledococele (divertículo intraduodenal). El tipo IV consta de dos subclasificaciones: en la IVa hay múltiples quistes intrahepáticos y extrahepáticos, y en la IVb, múltiples quistes extrahepáticos. Por último, el tipo V (enfermedad de Caroli) está constituido por quistes intrahepáticos múltiples.
Con respecto a la frecuencia de presentación, los artículos mencionan que en primer lugar está el tipo I, seguido por los tipos IV y V, finalmente, menos del 2 % de los casos corresponden a los tipos II y III 11.
Patogenia
La etiología de los quistes del colédoco aún no es clara. La teoría formulada por Babbitt en 1969 es la más popular 10. Se basa en la presencia de una anomalía en la unión bilio-pancreática, que consiste en la formación de un conducto largo común a ese nivel, el cual permite el reflujo del jugo pancreático hacia la vía biliar y la activación de las enzimas pancreáticas. A su vez, esto produce inflamación, denudación epitelial, adelgazamiento de la pared del conducto y evoluciona hacia la formación quística, con mayores presiones en el conducto pancreático que pueden seguir dilatando las paredes de la vía biliar afectada. A favor de esto, muchos estudios han demostrado que el nivel de amilasa en la bilis es mayor en los pacientes con quiste de colédoco, cuando se comparan con pacientes sanos 12. Además, los mayores niveles de amilasa se asocian significativamente con una menor edad de aparición de los síntomas y mayor grado de displasia.
La amilasa puede ser un marcador para el reflujo pancreático, pero es más probable que otras enzimas sean las causantes del daño epitelial, por lo que se han cuantificado el tripsinógeno y la fosfolipasa A2 en la bilis, cuyos niveles también se han encontrado elevados 13, 14. Se ha planteado que la enterocinasa del epitelio biliar alterado convierte el tripsinógeno en tripsina, la cual tiene efecto de irritante digestivo y activa la fosfolipasa A2; esta enzima activada hidroliza la lecitina y la convierte en liso-lecitina, que produce inflamación y ruptura de la pared biliar 15.
Sin embargo, se sabe que muchos pacientes con quistes del colédoco no poseen un canal biliopancreático común alargado, por lo que se ha intentado formular otras teorías para explicar su formación. Además, algunos pacientes con un conducto común largo no tienen quiste del colédoco asociado, por lo que esta observación no apoya la teoría del reflujo pancreático 4. Algunos partidarios de la teoría de Babbitt dicen que el canal común se considera largo según una longitud arbitraria, con una amplia variación en las medidas 16. De hecho, no es clara la definición de canal común largo, la cual cambia según el autor de 10 a 45 mm de longitud en cualquiera de sus porciones. Por lo tanto, un canal común largo puede presentarse en una proporción mucho mayor de pacientes con quistes del colédoco, pero puede ser subestimada debido a consideraciones poco realistas de la longitud del canal común o métodos inadecuados de imágenes.
La anomalía de la unión pancreato-biliar y el reflujo pancreato-biliar han cobrado mayor importancia en los últimos años, con el avance en el entendimiento de la fisiopatología y el aporte al cáncer de la vía biliar y la pancreatitis. La alteración de la unión pancreato-biliar se define como una anomalía congénita que consiste en la unión del conducto pancreático y vía biliar por fuera de la pared duodenal, formando un canal común y largo (>15 mm). De esta manera, el esfínter de Oddi no cumple su función y al existir una mayor presión hidrostática en el conducto pancreático, permite el reflujo a la vía biliar de jugo pancreático, principalmente, y de bilis; este produce daño endotelial e hiperplasia y metaplasia epiteliales, que promueven la progresión a carcinoma.
Actualmente, la colangiopancreatografía por resonancia magnética es muy importante en el diagnóstico, aunque pierde sensibilidad cuando se trata de canales comunes menores de 9 mm; en este caso, se utiliza la colangiografía directa. Comúnmente, este tipo de anormalidad cursa con unos niveles de amilasa en bilis muy elevados, pero en algunos casos no se puede encontrar ninguna elevación.
En un estudio japonés de 2.561 pacientes, se detectó neoplasia maligna en 21,6 % de aquellos con dilatación de la vía biliar y en 42,4 % de aquellos sin dilatación. En los primeros, la neoplasia asentó en la vía biliar en 31,2 % y en la vesícula biliar en 62,3 %; en los segundos, este porcentaje fue de 7,3% y 88,1 %, respectivamente. 17-19.
En la figura 2 se muestra la fisiopatología de la anomalía de la unión pancreato-biliar y del conducto común largo.
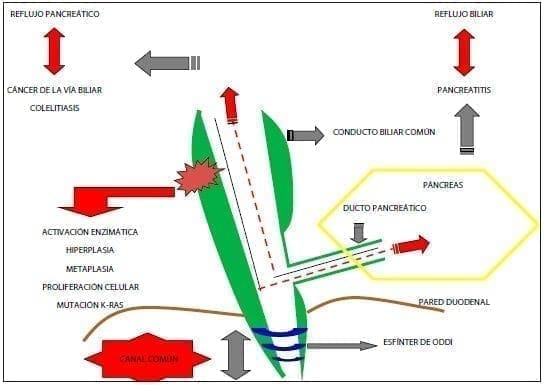 Figura 2. Fisiopatología del canal común y la mala unión biliopancreática.
Figura 2. Fisiopatología del canal común y la mala unión biliopancreática.
Kusunoki postuló otra teoría; este autor describió la reducción de células ganglionares en la pared del quiste, lo que correspondería a una oligoganglionosis biliar, similar a lo que se encuentra en la enfermedad de Hirschsprung del colon 12. En este caso, la inflamación crónica y los síntomas se producirían debido al flujo biliar lento en el lugar del reflujo pancreático 10.
Con las teorías anteriores se puede explicar la formación de los quistes de tipo I y de tipo IV, pero algunos autores sostienen que la etiología de los otros tipos es muy diferente. Los quistes de tipo II son verdaderos divertículos del conducto biliar común, con evidencia histológica de inflamación y potencial carcinogénico. La etiología de los coledococeles es discutida; Wheeler sugirió que la obstrucción de la ampolla de Vater puede dar lugar a una dilatación localizada de la vía biliar distal intramural 20. Los quistes de tipo V o enfermedad de Caroli, se deben a la detención en la remodelación y reabsorción selectiva de la placa del conducto, la cual comienza en la semana 12 del desarrollo de la gestación y progresa hasta formar los conductos biliares grandes en el hilio y los conductos pequeños en la periferia, lo cual se traduce en la malformación de la placa en los grandes conductos. La enfermedad de Caroli se asocia con atresia biliar, que también se relaciona con la malformación de la placa del conducto. Esta enfermedad también está asociada con una herencia autosómica recesiva y, con menor frecuencia, con el riñón poliquístico de presentación familiar 21-23. Se ha postulado que las mutaciones genéticas responsables de las malformaciones renales, también pueden dar lugar a malformaciones en la placa del conducto hepático 22.
Los quistes de colédoco se asocian con diferentes anomalías del desarrollo, que incluyen atresia de colon, atresia duodenal, ano no perforado, malformaciones arterio-venosas del páncreas, vesícula biliar multitabicada, defecto del tabique ventricular, hipoplasia de la aorta, páncreas divisum, aplasia pancreática, hiperplasia nodular focal, ausencia congénita de la vena porta, tejido pancreático heterotópico y poliposis familiar adenomatosa 24.
En el embrión, el páncreas se forma cuando rotan las yemas pancreáticas ventral y dorsal, se fusionan y forman conexiones con el árbol biliar; la rotación y fusión anormales pueden dar lugar a páncreas divisum, aplasia del páncreas, canal común largo o quistes del colédoco 25, 26.
Carcinogénesis
Actualmente, se acepta que los quistes del colédoco son un estado premaligno que produce mayor riesgo de cáncer (10 a 15 %), el cual aumenta con el tiempo, y también, adelanta su edad de aparición en 10 a 15 años, en promedio 611, 27 28.
En la tabla 1 se muestra la distribución de los tipos de cáncer en pacientes con quistes del colédoco 29.
El cáncer se localiza principalmente en el conducto biliar extrahepático (50 %) y la vesícula biliar (38 a 46 %), y en menor proporción, en la vía biliar intrahepática, el hígado y el páncreas 11. En una revisión, Todani, et al., encontraron que el 68 % de los casos de cáncer se asociaban con quistes de tipo I, el 5 %, con quistes de tipo II, el 1,6 %, con quistes de tipo III, el 21 %, con quistes de tipo IV, y el 6%, con quistes de tipo V 29. La unión anormal del conducto pancreático-biliar tiene un 16% al 55% de riesgo de malignidad con o sin la dilatación del conducto biliar 30. La enfermedad de Caroli se asocia con un riesgo de cáncer de 7 a 15 % 31. Generalmente, la incidencia de cáncer asociado con coledococele es de 2,5 %, pero en un estudio se reportó una incidencia alta, de 27 % 32.
Tabla 1.
Tipos de cáncer en pacientes con quistes del colédoco

1 Médico, residente de Cirugía General, Departamento de Cirugía, Universidad de Antioquia, Medellín, Colombia
2 Médica, residente de Pediatría, Universidad Militar Nueva Granada, Bogotá, D.C., Colombia
3 Médico cirujano, profesor titular, Facultad de Medicina, Universidad de Antioquia; Grupo de Gastrohepatología, Universidad de Antioquia; Unidad de Cirugía Hepatobiliar y Pancreática Hospital Pablo Tobón Uribe; Grupo de Trasplante Hepático, Hospital Pablo Tobón Uribe y Universidad de Antioquia, Medellín, Colombia
Fecha de recibido: 16 de enero de 2015
Fecha de aprobación: 19 de agosto de 2015
Citar como: Sánchez JA, Gómez S, Morales C, Hoyos SI. Quistes del colédoco. Rev Colomb Cir. 2015;30:296-305.
Diagnóstico
En 80 % de los casos los quistes del colédoco se manifiestan clínicamente antes de los 10 años, aunque pueden hacerlo a cualquier edad. La tríada clásica de síntomas, que consiste en dolor abdominal, ictericia y masa abdominal palpable, se produce en menos de 20 % de los pacientes, aunque casi en las dos terceras partes de los casos se presentan dos de estos tres síntomas 33.
Los síntomas son diferentes en pacientes menores de 12 meses y en mayores de 12 meses. Los menores de un año suelen presentar ictericia obstructiva y masa abdominal, mientras que, en aquellos con mayor edad, son más frecuentes el dolor abdominal, la fiebre, las náuseas, el vómito y la ictericia 22, 34, 35.
Las complicaciones de los quistes del colédoco son resultado de la estasis biliar, la cual facilita la formación de cálculos, la sobreinfección recurrente y la inflamación. La obstrucción y las infecciones, especialmente en pacientes con afectación intrahepática, llevan a cirrosis biliar secundaria, de tal manera que también se pueden presentar signos y síntomas de hipertensión portal, como hemorragia de vías digestivas altas, esplenomegalia y pancitopenia.
Entre 1 y 12 % de los pacientes presentan ruptura espontánea del quiste con dolor abdominal, sepsis y peritonitis 36; en estos casos, el diagnóstico se hace por presencia de líquido bilioso en la paracentesis, o de fuga de medio de contraste peritoneal, observada en la gammagrafía hepatobiliar con ácido iminodiacético (HIDA) 32.
El examen inicial para la valoración de la vía biliar debe ser una ecografía, la cual sugiere el diagnóstico de quistes del colédoco en la mayoría de los pacientes; no obstante, la resonancia magnética (RM) con contraste es el estudio de elección para valorar estas estructuras 37. Es importante tener en cuenta que algunos casos son difíciles de diagnosticar, incluso contando con la tecnología para ello, 38 ya que el aire, la sangre, los cálculos o los tapones de proteínas intraductales, comunes en pacientes con quistes del colédoco, pueden interferir con la señal y alterar la visualización 39. Sin embargo, la sensibilidad de la colangiorresonancia magnética para el diagnóstico es cercana al 100 % 6. La colangiopancreatografía por resonancia magnética tiene una sensibilidad de 84 % para obtener imágenes de anastomosis posoperatorias, pero su sensibilidad para evaluar la unión pancreático-biliar es tan baja como 46 % a 60 % 36.
Las imágenes por resonancia magnética no son buenas para visualizar cálculos menores de 5 mm en los conductos o conductos tortuosos 37. Algunos autores sugieren que la baja sensibilidad de la RM en la visualización de la salida pancreático-biliar se relaciona con el pequeño calibre de esta unión y sugieren la administración de secretina antes de obtener las imágenes, lo que aumenta la secreción pancreática y dilata los conductos para su mejor visualización 40. Las ventajas de la RM sobre la colangiopancreatografía retrógrada endoscópica son que no se usa radiación ionizante, no es invasiva, no depende del operador y no produce complicaciones como colangitis o pancreatitis 37.. La segunda permite la intervención terapéutica, pero esta solo es necesaria en los quistes del colédoco de tipo III 39.
El diagnóstico diferencial de los quistes de tipo III incluye los divertículos duodenales y los quistes de duplicación. Los divertículos se llenan de contraste en las series gastrointestinales superiores y no presentan opacidad en la colangiopancreatografía retrógrada endoscópica. Los quistes de duplicación producen imágenes idénticas a las de los coledococeles y son muy difíciles de diferenciar. Algunos autores afirman que en los quistes de duplicación existe pared muscular y en los coledococeles no 41.
El diagnóstico diferencial de la enfermedad de Caroli incluye colangitis piógena recurrente, enfermedad hepática poliquística y colangitis esclerosante primaria. Bloustein describió el “signo del punto central”, que representa la protrusión intraluminal del paquete fibrovascular formado por las pequeñas ramas de la vena porta y la arteria hepática, y es una imagen característica en resonancia y tomografía, aunque no patognomónica de la enfermedad de Caroli, ya que también se puede ver en las dilataciones obstructivas 37,42. Esto sugiere que la enfermedad de Caroli es intraductal y consiste en tabiques ecogénicos que atraviesan los conductos 36.
La colangitis piógena recurrente se manifiesta como dilataciones no saculares intrahepáticas y extrahepáticas, con cálculos que llenan la luz. En la enfermedad hepática poliquística, los quistes no se comunican con el árbol biliar. La colangitis esclerosante primaria se manifiesta por dilataciones leves, focales y fusiformes, con obstrucción distal evidente, y frecuentemente, se asocia con enfermedad inflamatoria intestinal. Estas características pueden ayudar a diferenciar la enfermedad de Caroli de otras.
Tratamiento
La resección quirúrgica completa con colecistectomía es el tratamiento de elección de los quistes del colédoco 3,40, requiriéndose reconstrucción de la vía biliar sobre el remanente del conducto biliar proximal sano con hepatoyeyunostomía en “Y” de Roux , excepto en quistes de tipo III que la aproximación endoscópica es la inicial 43-45. Las siguientes son las técnicas quirúrgicas utilizadas según el tipo de quistes:
Tipo I: requieren colecistectomía y resección completa de los conductos extrahepáticos y reconstrucción mediante hepático-yeyunostomía en “Y” de Roux.
Tipo II: requieren resección del quiste y colecistectomía; la resección completa de las vías biliares extrahepáticas no es necesaria. Según el tamaño del cuello del quiste, se pueden utilizar el cierre primario o el tubo de tipo Kehr.
Tipo III (coledococele): cuando el quiste es menor de 3 cm, la esfinterotomía endoscópica es efectiva, debido a que el riesgo de neoplasia maligna en este tipo de quistes es mucho menor; aquellos mayores de 3 cm, que pueden producir obstrucción, requieren escisión transduodenal y, ocasionalmente, reimplante del conducto pancreático en la pared duodenal.
Tipo IVa: generalmente, el único tratamiento es la resección y la reconstrucción mediante hepático-yeyunostomía; si las lesiones intrahepáticas están restringidas a un solo lóbulo hepático, se necesita una hepatectomía parcial.
Tipo IVb: requiere resección completa de la porción comprometida de la vía biliar extrahepática, con escisión transduodenal o esfinterotomía endoscópica
Tipo V (enfermedad de Caroli): si las lesiones están limitadas a un lóbulo (generalmente, el izquierdo), es suficiente la hepatectomía parcial con colangio-yeyunostomía, según el caso.
Algunos pacientes con quistes que no se pueden resecar, especialmente intrahepáticos, o con enfermedad bilobular, pueden requerir trasplante hepático.
En general, cuando existe compromiso intrahepático debe prestarse especial atención a la posibilidad de hepatolitiasis. La funcionalidad y la reserva hepáticas siempre deben tenerse en cuenta, antes de cualquier resección hepática.
Cuando el quiste se adhiere a la vena porta por inflamación crónica grave, se puede utilizar la técnica de Lilly; esta consiste en dejar la porción serosa adherida a la vena porta, sin intentar la resección completa, y legrar la mucosa o intentar cauterizarla de manera cuidadosa.
Es importante destacar que la incidencia de complicaciones es menor cuando el tratamiento inicial es la resección total del quiste, ya que se minimizan los riesgos de complicaciones y de transformación maligna 4. Algunos casos de quistes de tipo III, principalmente los pequeños, pueden beneficiarse del tratamiento endoscópico con esfinterotomía, ya que tienen un riesgo bajo de transformación maligna, aunque se debe hacer un seguimiento estrecho.
Son frecuentes las complicaciones quirúrgicas e incluyen resección incompleta, cálculos intrahepáticos, colangitis, pancreatitis posoperatoria, fístula biliar y reintervención. De estas, la más usual es la resección parcial del quiste con quistoenterostomía, la cual, además, genera con mayor frecuencia colangitis y cáncer metacrónico. Por lo anterior, debe propenderse por la resección completa de los quistes, sin olvidar que algunos comprometen proximalmente la vía biliar en la unión de los conductos hepáticos; esto se presenta en cerca de 14 % de los casos de quistes de tipo I o IV, hasta en 86% de los cuales puede haber resección incompleta, con una incidencia aproximada de cáncer de 29 % 46. Las estenosis de las anastomosis posoperatorias también son complicaciones frecuentes y están asociadas a litiasis intrahepática y colangitis 47; la incidencia de las estenosis se reduce cuando se hace una anastomosis más alta 48.
La cirugía laparoscópica se utiliza cada vez más y trae beneficios como menor trauma quirúrgico, menor sangrado, mejor resultado estético, menor infección del sitio operatorio, mejor analgesia posoperatoria y recuperación posoperatoria más rápida. En los quistes de colédoco también se aplica esta técnica, en los cuales se ha descrito la resección laparoscópica con hepático-yeyunostomía, la cual parece ser segura y eficaz, e incluso, podría convertirse en el método ideal de tratamiento, especialmente en niños 49. Por su seguridad y bajo riesgo, la cirugía laparoscópica puede ser el tratamiento de elección para los quistes de colédoco de tipo II 48. Incluso, ya hay reportes de casos de la aplicación de cirugía robótica para resecar quistes de colédoco 50, pero, para que se convierta en el método de elección, hace falta practicarla más y mejores estudios 48, 51, 52.
La cirugía debe practicarse lo más pronto posible después del diagnóstico, para evitar complicaciones y, en especial, para evitar el daño hepático en neonatos 53. Con el avance de la ecografía y los mejores programas de control prenatal, los quistes de colédoco se diagnostican con mayor frecuencia desde la etapa prenatal. Usualmente, se postula que la intervención debe practicarse idealmente a los tres a seis meses de edad; no obstante, en estos pacientes se presenta fibrosis hepática temprana que progresa rápidamente, por lo cual, aunque se encuentren asintomáticos, deben someterse a cirugía, idealmente en el periodo neonatal 54.
En casos de lesiones benignas, puede considerarse el trasplante hepático cuando existe compromiso extenso del parénquima hepático, hay complicaciones que comprometan la vida o si no puede descartarse enfermedad maligna. Se ha reportado en pequeñas series de casos de enfermedad de Caroli y no hay guías claras disponibles. En los casos y series de casos informados, se refieren supervivencias a cinco años del paciente o el injerto entre 70 y 86 %, con alta incidencia de complicaciones posoperatorias tempranas, como sepsis. Aunque a largo plazo los resultados son buenos, aún hay controversia sobre esta opción de tratamiento 55.
Riesgo de transformación maligna después de la resección
En una serie con búsqueda bibliográfica exhaustiva, el porcentaje de transformación maligna después de la resección fue de 0,7 % 56. Esta se puede presentar en la porción residual intrapancreática del quiste después de la resección 56; su reconocimiento ha hecho que se opte por la resección de la porción intrapancreática del conducto biliar común 57.
La amplitud de la resección en los quistes de tipo IVa aún es controversial. Aunque algunos autores recomiendan la resección del componente extrahepático solamente con hepático-enterostomía 7, se ha reportado carcinogenia a partir del componente intrahepático, así como también después de la resección completa 58. Por lo tanto, cuando la enfermedad se encuentre localizada y no dispersa en el hígado, se recomienda la resección del lóbulo hepático correspondiente 53,59,60.
El riesgo de transformación maligna después de resecar los quistes de colédoco, es relativamente alto a largo plazo, con aparición de cáncer metacrónico en 9,9 % de los casos 11. Además, el riesgo de neoplasia biliar maligna en el conducto biliar remanente, continúa aumentando con el tiempo: es de 1,6 % a los 15 años, de 3,9 % a los 20 años, y de 11,3 % a los 25 años 61. Los factores de riesgo para recurrencia, son: edad mayor de 40 años, ausencia de vesícula biliar, elevación del antígeno carcinoembrionario o del biomarcador CA 19-9, y anomalía de la unión pancreático-biliar. Por lo tanto, es necesario un estrecho seguimiento después de la resección 11, debido al mal pronóstico en los casos de recurrencia 61.
Conclusiones
Los quistes del colédoco son una entidad rara y tienen una presentación clínica muy variada, por lo cual se requiere un índice de sospecha alto para evitar retrasos en el diagnóstico y complicaciones. El método imaginológico que se debe utilizar es la colangiorresonancia, la cual permite una caracterización adecuada y facilita la planeación quirúrgica. Se debe practicar resección completa del quiste y colecistectomía, mediante técnica abierta o laparoscópica, pues así se disminuye el riesgo de complicaciones y de degeneración maligna. Para los quistes de colédoco de tipo III es suficiente el tratamiento mediante esfinterotomía endoscópica, pero siempre se requiere el se guimiento a largo plazo.
Agradecimientos
Proyecto Sostenibilidad, Vicerrectoría de Investigación, Universidad de Antioquia
Choledochal cysts
Abstract
Choledochal cysts are rare pathological conditions characterized by dilatation of the intrahepatic and extrahepatic bile ducts. Postulated etiologies are an abnormal pancreato-biliary junction favoring reflux of pancreatic juice to the biliary tree, and aganglionosis of the biliary tree. They have a wide range of presenting symptoms, the main ones being abdominal pain, jaundice and evident abdominal mass. Choledochal cysts are premalignant lesions, and therefore the treatment of choice is complete surgical resection, which demands long-term follow-up.
Key words: Common bile duct; choledochal cyst; ampulla of Vater; biliary tract neoplasms; anastomosis, surgical.
Bibliografía
1. Mcwhorter GL. Congenital Cystic Dilatation Of The Com¬mon Bile Duct: Report Of A Case, With Cure. Arch Surg. 1924;8:604-26.
2. O’Neill Jr JA. Choledochal Cyst. Curr Prob Surg. 1992;29:361- 410.
3. Powell CS, Sawyers JL, Reynolds VH. Management Of Adult Choledochal Cysts. Ann Surg. 1981;193:666.
4. Nagorney D, Mcilrath D, Adson M. Choledochal Cysts In Adults: Clinical Management. Surgery. 1984;96:656-63.
5. Ono J, Sakoda K, Akita H. Surgical Aspect Ot Cystic Dilatation Of The Bile Duct. An Anomalous Junction Of The Pancreatico¬biliary Tract In Adults. Ann Surg. 1982;195:203.
6. Gigot J, Nagorney D, Farnell M, Moir C, Ilstrup D. Bile Duct Cysts: A Changing Spectrum Of Presentation. J Hepatobiliary Pancreat Surg. 1996;3:405-11.
7. López RR, Pinson CW, Campbell JR, Harrison M, Katon RM. Variation In Management Based On Type Of Choledochal Cyst. Am J Surg. 1991;161:612-5.
8. Chijiiwa K, Koga A. Surgical Management And Long-Term Follow-Up Of Patients With Choledochal Cysts. Am J Surg. 1993;165:238-42.
9. Alonso-Lej F, Rever WB Jr, Pessagno DJ, Congenital chole¬dochal cyst, with a report of 2, and an analysis of 94, cases Int Abstr Surg. 1959;108:1-30.
10. Todani T, Watanabe Y, Narusue M, Tabuchi K, Okajima K. Congenital bile duct cysts: Classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. Am J Surg. 1977;134:263-9.
11. Lee SE, Jang JY, Lee YJ, Choi DW, Lee WJ, Cho BH, et al. Choledochal cyst and associated malignant tumors in adults: A multicenter survey in South Korea. Arch Surg. 2011;146: 1178-84.
12. Sugiyama M, Haradome H, Takahara T, Izumisato Y, Abe N, Masaki T, et al. Biliopancreatic reflux via anomalous pancrea¬ticobiliary junction. Surgery. 2004;135:457-9.
13. Ochiai K, Kaneko K, Kitagawa M, Ando H, Hayakawa T. Activa¬ted pancreatic enzyme and pancreatic stone protein (PSP/reg) in
bile of patients with pancreaticobiliary maljunction/choledochal cysts. Dig Dis Sci. 2004;49:1953-6.
14. Narita H, Hashimoto T, Suzuki T, Kamiya Y, Murata Y, Hayashi S, et al. Clinical and experimental studies on the activation me¬chanism of pancreatic enzymes refluxing into the biliary tract with an anomalous pancreatico-biliary ductal junction. Nippon Shonika Gakkai Zasshi. 1990;26:609-15.
15. Mizuno M, Kato T, Koyama K. An analysis of mutagens in the contents of the biliary tract in pancreaticobiliary maljunction. Surg Today. 1996;26:597-602.
16. Davenport M, Stringer M, Howard E. Biliary amylase and con¬genital choledochal dilatation. J Pediatr Surg. 1995;30:474-7.
17. Kamisawa T, Kuruma S, Tabata T, Chiba K,Iwasaki S, Koizumi S, Pancreaticobiliary maljunction and biliary cáncer, J Gastroen¬terol. 2015;50:273-9..
18. Kamisawa T, Anjiki H, Egawa N, Kurata M, Honda G, Tsuruta K, Diagnosis and clinical implications of pancreatobiliary reflux. World J Gastroenterol. 2008;14:6622-6
19. Horaguchi J, Fujita N, Kamisawa T, Honda G, Chijiiwa K, Ma¬guchi H, Tanaka M, Pancreatobiliary reflux in individuals with a normal pancreaticobiliary junction: a prospective multicenter study. J Gastroenterol. 2014;49:875-81.
20. de Courcy Wheeler WI. An unusual case of obstruction to the common bile-duct (choledochocele?) Br J Surg 1940;27:446-8. Article first published online: 6 Dec 2005
21. Mousson C, Rabec M, Cercueil J, Virot J, Hillon P, Rifle G. Caroli’s disease and autosomal dominant polycystic kid¬ney disease: A rare association? Nephrol Dial Transplant. 1997;12:1481-3.
22. Ninan VT, Nampoory MR, Johny KV, Gupta RK, Schmidt I, Nair PM, et al. Caroli’s disease of the liver in a renal transplant recipient. Nephrol Dial Transplant. 2002;17:1113-5.
23. Yonem O, Bayraktar Y. Clinical characteristics of Caroli’s syn¬drome. World J Gastroenterol. 2007;13:1934-7.
24. Elton E, Hanson BL, Biber BP, Howell DA. Dilated common channel syndrome: Endoscopic diagnosis, treatment, and re¬lationship to choledochocele formation. Gastrointest Endosc. 1998;47:471-8.
25. Hosoki T, Hasuike Y, Takeda Y, Michita T, Watanabe Y, Sakamori R, et al. Visualization of pancreaticobiliary reflux in anomalous pancreaticobiliary junction by secretin-stimulated dynamic magnetic resonance cholangiopancreatography. Acta Radiol. 2004;45:375-82.
26. Kinjo T, Aoki H, Sunagawa H, Kinjo S, Muto Y. Congenital absence of the portal vein associated with focal nodular hyper¬plasia of the liver and congenital choledochal cyst: A case report. J Pediatr Surg. 2001;36:622-5.
27. Tsuchiya R, Harada N, Ito T, Furukawa M, Yoshihiro I, Kusano T, et al. Malignant tumors in choledochal cysts. Ann Surg. 1977;186:22-8.
28. Bismuth H, Krissat J. Choledochal cystic malignancies. Ann Oncol. 1999;10(Suppl.4):S94-8.
29. Chaudhary A, Dhar P, Sachdev A, Kumar N, Vij J, Sarin S, et al. Choledochal cysts differences in children and adults. Br J Surg. 1996;83:186-8.
30. Lee HC, Yeung CY, Fang SB, Jiang CB, Sheu JC, Wang NL. Biliary cysts in children–long-term follow-up in Taiwan. J Formos Med Assoc. 2006;105:118-24.
31. O’Neill Jr JA, Templeton Jr JM, Schnaufer L, Bishop HC, Ziegler MM, Ross 3rd A. Recent experience with choledochal cyst. Ann Surg. 1987;205:533.
32. Moss RL, Musemeche CA. Successful management of ruptured choledochal cyst by primary cyst excision and biliary recons¬truction. J Pediatr Surg. 1997;32:1490-1.
33. Shi L-B, Peng S-Y, Meng X-K, Peng C-H, Liu Y-B, Chen X-P, et al. Diagnosis and treatment of congenital choledochal cyst: 20 years experience in China. World J Gastroenterol. 2001;7:732-4.
34. Visser BC, Suh I, Way LW, Kang SM. Congenital choledochal cysts in adults. Arch Surg. 2004;139:855-62.
35. Germani M, Liberto D, Elmo G, Lobos P, Ruiz E. Choledochal cyst in pediatric patients: A 10 years single institution experience. Acta Gastroenterol Latinoam. 2011;41:302-7.
36. Lam WW, Lam TP, Saing H, Chan FL, Chan KL. MR cholan¬giography and CT cholangiography of pediatric patients with choledochal cysts. AJR Am J Roentgenol. 1999;173:401-5.
37. Hussain SZ, Bloom DA, Tolia V. Caroli’s disease diagnosed in a child by MRCP. Clin Imaging. 2000;24:289-91.
38. Arrieta A, Manzano A, Navarro D, Durango R. Dilema en el diagnóstico de quiste de colédoco: reporte de un caso. Gen. 2011;65:237-9.
39. Arshanskiy Y, Vyas PK. Type IV choledochal cyst presenting with obstructive jaundice: Role of MR cholangiopancreato¬graphy in preoperative evaluation. AJR Am J Roentgenol. 1998;171:457-9.
40. Matos C, Nicaise N, Devière J, Cassart M, Metens T, Struyven J, et al. Choledochal cysts: Comparison of findings at MR cho-langiopancreatography and endoscopic retrograde cholangio¬pancreatography in eight patients. Radiology. 1998;209:443-8.
41. Arenas-Jiménez JJ1, Gómez-Fernández-Montes J, Mas-Estellés F, Cortina-Orts H. Large choledochocoele: difficulties in radio¬logical diagnosis. Pediatr Radiol. 1999;29:807-10.
42. Purandare D, Thakkar H, Lolge S, Purandare N. Intraluminal portal vein sign in Caroli’s syndrome. Indian J Gastroenterol. 2004;23:158.
43. Scudamore CH, Hemming AW, Teare JP, Fache JS, Erb SR, Watkinson AF. Surgical management of choledochal cysts. Am J Surg. 1994;167:497-500.
44. Stain SC, Guthrie CR, Yellin AE, Donovan AJ. Choledochal cyst in the adult. Ann Surg. 1995;222:128.
45. Martin RF, Biber BP, Bosco JJ, Howell DA. Symptomatic choledochoceles in adults: Endoscopic retrograde cholangio¬
pancreatography recognition and management. Arch Surg. 1992;127:536-9.
46. Mabrut JY, Partensky C, Gouillat C, Baulieux J, Ducerf C, Kes¬tens PJ, et al. Cystic involvement of the roof of the main biliary convergence in adult patients with congenital bile duct cysts: A difficult surgical challenge. Surgery. 2007;141:187-95.
47. Ando H, Ito T, Kaneko K, Seo T, Ito F. Intrahepatic bile duct stenosis causing intrahepatic calculi formation following exci¬sion of a choledochal cyst. J Am Coll Surg. 1996;183:56-60.
48. Liu DC, Rodríguez JA, Meric F, Geiger JL. Laparoscopic exci¬sion of a rare type II choledochal cyst: Case report and review of the literature. J Pediatr Surg. 2000;35:1117-9.
49. Wang B, Feng Q, Mao JX, Liu L, Wong KK. Early experience with laparoscopic excision of choledochal cyst in 41 children. J Pediatr Surg. 2012;47:2175-8.
50. Sánchez A, Rodríguez O, Peña R, Salamo O, Sosa E, Dávila H. Cirugía robótica: resección de quiste de colédoco tipo I y hepaticoyeyunoanastomosis en Y de Roux. Reporte de un caso. Revista Vitae Academia Biomédica Digital. 2012. Fecha de con¬sulta: 20 de agosto de 2014. Disponible en https://www.bioline. org.br/www.bioline.org.br/titles?id=va&year=2012&vol=0&n um=49&keys=V0N49
51. Tanaka M, Shimizu S, Mizumoto K, Yokohata K, Chijiiwa K, Yamaguchi K, et al. Laparoscopically assisted resection of choledochal cyst and Roux-en-Y reconstruction. Surg Endosc. 2001;15:545-51.
52. Shimura H, Tanaka M, Shimizu S, Mizumoto K. Laparosco¬pic treatment of congenital choledochal cyst. Surg Endosc. 1998;12:1268-71.
53. Shito M, Shintoku J, Miyazaki H, Mukai M. Asymptomatic intrahepatic choledochal cyst associated with chronic active hepatitis C. Hepatogastroenterology. 1997;45:2356-8.
54. Diao M, Li L, Cheng W. Timing of surgery for prenatally diagno¬sed asymptomatic choledochal cysts: A prospective randomized study. J Pediatr Surg. 2012;47:506-12.
55. Ercolani G, Grazi GL, Pinna AD. Liver transplantation for benign hepatic tumors: A systematic review. Dig Surg. 2010;27:68-75.
56. Watanabe Y, Toki A, Todani T. Bile duct cancer developed after cyst excision for choledochal cyst. J Hepatobiliary Pancreat Surg. 1999;6:207-12.
57. Ando H, Kaneko K, Ito T, Watanabe Y, Seo T, Harada T, et al. Complete excision of the intrapancreatic portion of choledochal cysts. J Am Coll Surg. 1996;183:317-21.
58. Kobayashi S, Asano T, Yamasaki M, Kenmochi T, Nakagohri T, Ochiai T. Risk of bile duct carcinogenesis after excision of ex¬trahepatic bile ducts in pancreaticobiliary maljunction. Surgery. 1999;126:939-44.
59. Nakayama H, Masuda H, Ugajin W, Koshinaga T, Fukuzawa M. Left hepatic lobectomy for type IV-A choledochal cyst. Am Surg. 2000;66:1020-2.
60. Granero LE, Marinelli P, Andrada DG, Marangoni A, Casaretto E. Quiste de coledoco tipo IV A. Reporte de un caso. Acta Gas¬troenterol Latinoam. 2009;39:193-6.
61. Ohashi T, Wakai T, Kubota M, Matsuda Y, Arai Y, Ohyama T, et al. Risk of subsequent biliary malignancy in patients undergoing cyst excision for congenital choledochal cysts. J Gastroenterol Hepatol. 2013;28:243-7.
Correspondencia: Sergio Iván Hoyos, MD
Correo electrónico: sergiohoyosd@yahoo.es
Medellín, Colombia