Síndrome de Inmunodeficiencia Severa Combinada (IDSC)
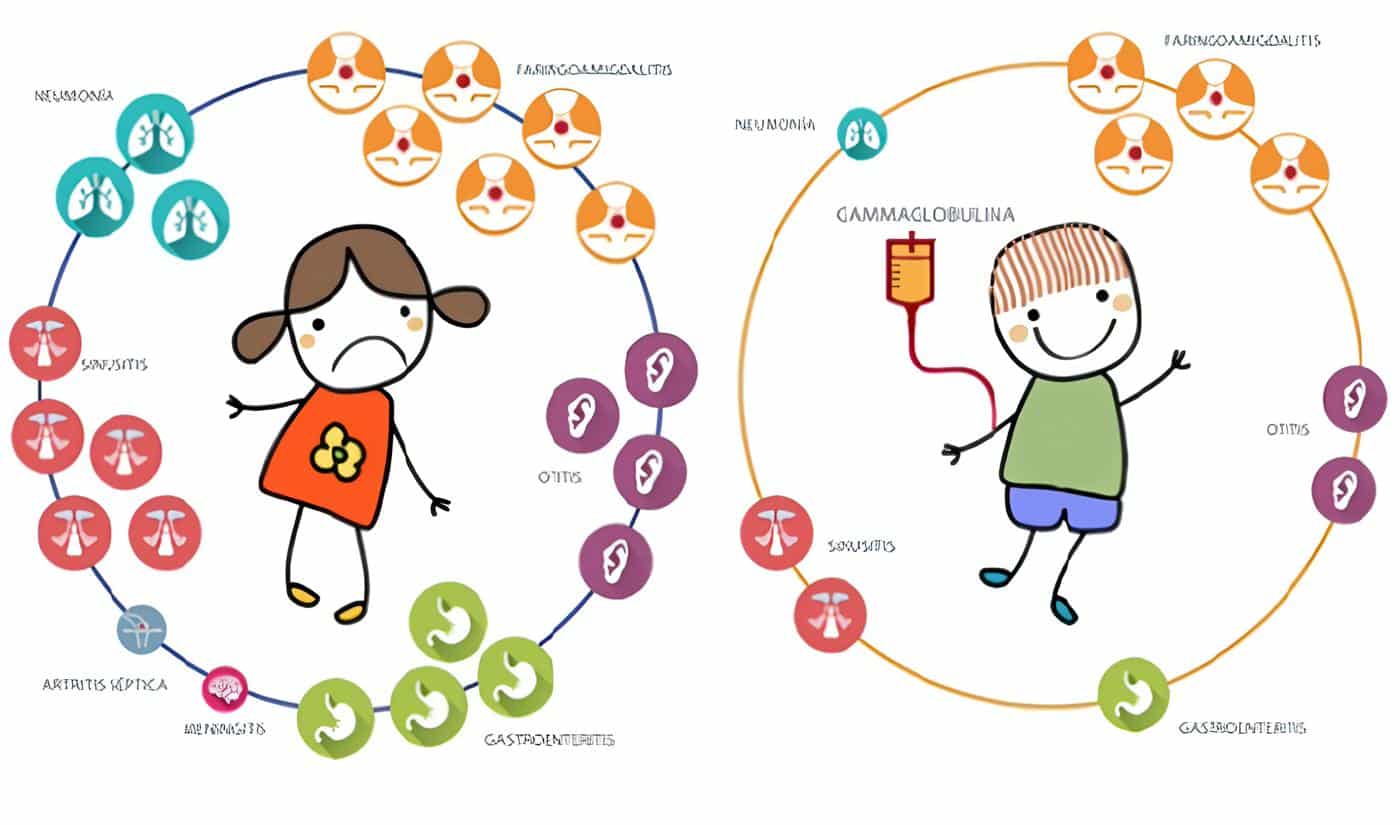
La IDSC representa la forma más severa de las IDP. Es un síndrome que agrupa varias enfermedades de origen genético diverso que comparten la característica de un bloqueo profundo en la diferenciación y/o función de los linfocitos T y B. En la actualidad se reconocen ocho formas diferentes y se defininen de acuerdo al patrón de herencia, el fenotipo y la identificación de los genes mutados.
El diagnóstico de cada enfermedad se orienta con los diferentes estudios en el laboratorio (LT CD3+, CD4+ y CD8+; linfocitos B (LB) CD19+; células NK CD16/CD56+) y se confirma investigando a nivel molecular el daño en las respectivas proteínas (1). Sin embargo, en el 20% de los casos de IDSC el defecto genético molecular permanece desconocido (12).
La IDSC se clasifica en tres grupos con base en el fenotipo de los linfocitos circulantes y el patrón de herencia encontrado (13):
1. IDSC T(-)B(-):
Con disminución notable en los LT y B; afecta por igual a ambos sexos y tiene un patrón de herencia autosómico recesivo. Las células NK pueden encontrarse en número normal o estar elevadas. Las entidades caracterizadas en este grupo son la deficiencia total de las enzimas recombinasas RAG1 y RAG2, la disgenesia reticular y la deficiencia de adenosina deaminasa (ADA).
2. IDSC T(-)B(+):
Con bajo número de LT pero normal o elevados los LB; las células NK por lo general están ausentes. De acuerdo con el patrón de herencia se encuentran dos clases de IDSC T(-)B(+) que comparten fenotipo y expresión clínica, la deficiencia de la cadena gama común del receptor de IL-2, ligada al cromosoma X (50% de las IDSC), y la deficiencia de la tirosina quinasa JAK3, autosómica recesiva.
3. IDSC T(+)B(-):
En este grupo se ubica el Síndrome de Omenn, una deficiencia parcial de las proteínas del sistema activador de las recombinasas RAG1 y RAG2; los LT circulantes son normales pero presentan restricción en la expresión de las cadenas a y b del receptor de los LT (TCR).
El Síndrome de Omenn es una entidad transmitida como un rasgo autosómico recesivo caracterizado por una eritrodermia exudativa generalizada.
Se manifiesta desde las primeras semanas de vida con alopecia, fiebre, diarrea prolongada, edema por pérdida de proteínas, linfadenopatía, hepatoesplenomegalia e infecciones severas de piel y pulmones, además de sepsis. En los órganos comprometidos existe infiltración por linfocitos, histiocitos y eosinófilos (14) (Foto 4).
A pesar de su etiología variable, existen hallazgos comunes en los diferentes tipos de IDSC: trastornos en el desarrollo del timo, linfopenia de leve a marcada, anormalidad en el desarrollo pondoestatural, susceptibilidad elevada a las infecciones recurrentes severas, candidiasis mucocutánea recurrente, diarrea persistente y neumonía intratable por Pneumocystis carinii (15).
Se observan otras manifestaciones cutáneas en la IDSC: candidiasis mucocutánea de aparición temprana, resistente a la terapia antimicótica y con tendencia a la cronicidad; dermatitis en el área del pañal y lesiones similares a una dermatitis seborreica; además presentan en forma temprana exantema cutáneo debido a una enfermedad de injerto contra hospedero, originada por los linfocitos T maternos que pasan a la circulación fetal al final de la gestación, o secundaria a una transfusión de sangre no irradiada (16).
(Lea También: Síndrome de Chediak-Higashi (SCH))
Abscesos cutáneos
Deficiencia de Adhesión de Leucocitos Tipo I (DAL-I)
La DAL-I es una enfermedad autosómica recesiva debida a mutaciones en el gen que codifica para la proteína CD18, la cadena b de las Beta-2 integrinas leucocitarias LFA-1, MAC-1 y p150,95. La LFA-1 y el MAC-1 son moléculas de adhesión importantes para la interacción firme de los leucocitos con el endotelio activado y la migración a través de la matriz extracelular hacia los sitios de inflamación.
El MAC-1 es un receptor leucocitario para péptidos derivados de la activación del complemento (CR3) que facilita la fagocitosis de los microorganismos opsonizados.
Por una expresión deficiente de estas integrinas leucocitarias, las células fagocíticas no migran hacia el foco inflamatorio para realizar la reacción microbicida, por lo cual se acumulan en la sangre periférica, causando una leucocitosis severa, en ausencia de infección (17, 18).

Foto 4. Síndrome de Omenn. Eritrodermia descamativa
Las alteraciones de la piel se inician en el recién nacido, con retardo en la caída del cordón umbilical, onfalitis y celulitis periumbilical por cicatrización lenta y deficiente. Por la ausencia de fagocitos, se presentan úlceras necróticas sin supuración purulenta. Las infecciones en las cavidades profundas se fistulizan rápidamente a la piel. Además, se observan gingivoestomatitis crónica y periodontitis (16, 17) (Foto 5).

Foto 5. Deficiencia de adhesión de leucocitos Tipo I. Onfalitis y celulitis severa.
Petequias y/o púrpura
Síndrome de Wiskott Aldrich (SWA)
El SWA se hereda con un patrón recesivo ligado al cromosoma X y se origina por mutaciones en un gen que codifica para una proteína denominada Proteína del Síndrome de Wiskott Aldrich (WASP), que tiene homología con las proteínas que se unen a la actina del citoesqueleto y participan en la reorganización del citoesqueleto de los leucocitos y las plaquetas.
La WASP sólo se expresa en los leucocitos y megacariocitos, esto explica la restricción de los síntomas a los sistemas inmune y de la coagulación (19).
En el SWA hay defectos en el número y la función de los LT y las plaquetas. Los LT tienen anormalidades en su capacidad de división, movimiento y organización de los receptores antigénicos por alteraciones en la reorientación del citoesqueleto; los LT CD8+ tienen disminución en su actividad citotóxica por la incapacidad para atacar las células blanco.
La función de los LB y los fagocitos son aparentemente normales; sin embargo, hay defectos en la interacción entre las células T y B que llevan a una deficiente producción de anticuerpos específicos, especialmente contra los antígenos del tipo polisacáridos (8).
Las anormalidades plaquetarias son un sello característico del SWA; se presenta trombocitopenia por defectos en su producción y por una elevada destrucción en el bazo. En consecuencia, desde los primeros meses de vida están presentes los sangrados por piel y mucosas con petequias, equimosis y hematoquezia. Un rasgo específico de esta enfermedad es la presencia de microtrombocitos (Foto 6).

Foto 6. Síndrome de Wiskott Aldrich. Hemorragia subconjuntival y equimosis en el párpado superior.
La manifestación dermatológica más característica del SWA en el 80% de los pacientes es un eczema indistinguible de la dermatitis atópica.
Aparece en los primeros meses de vida, con evolución crónica y en algunos casos, muy severa. Compromete la cara, el cuero cabelludo y las áreas de flexión. Con frecuencia, una infección bacteriana secundaria coloniza las lesiones eczematosas (16).
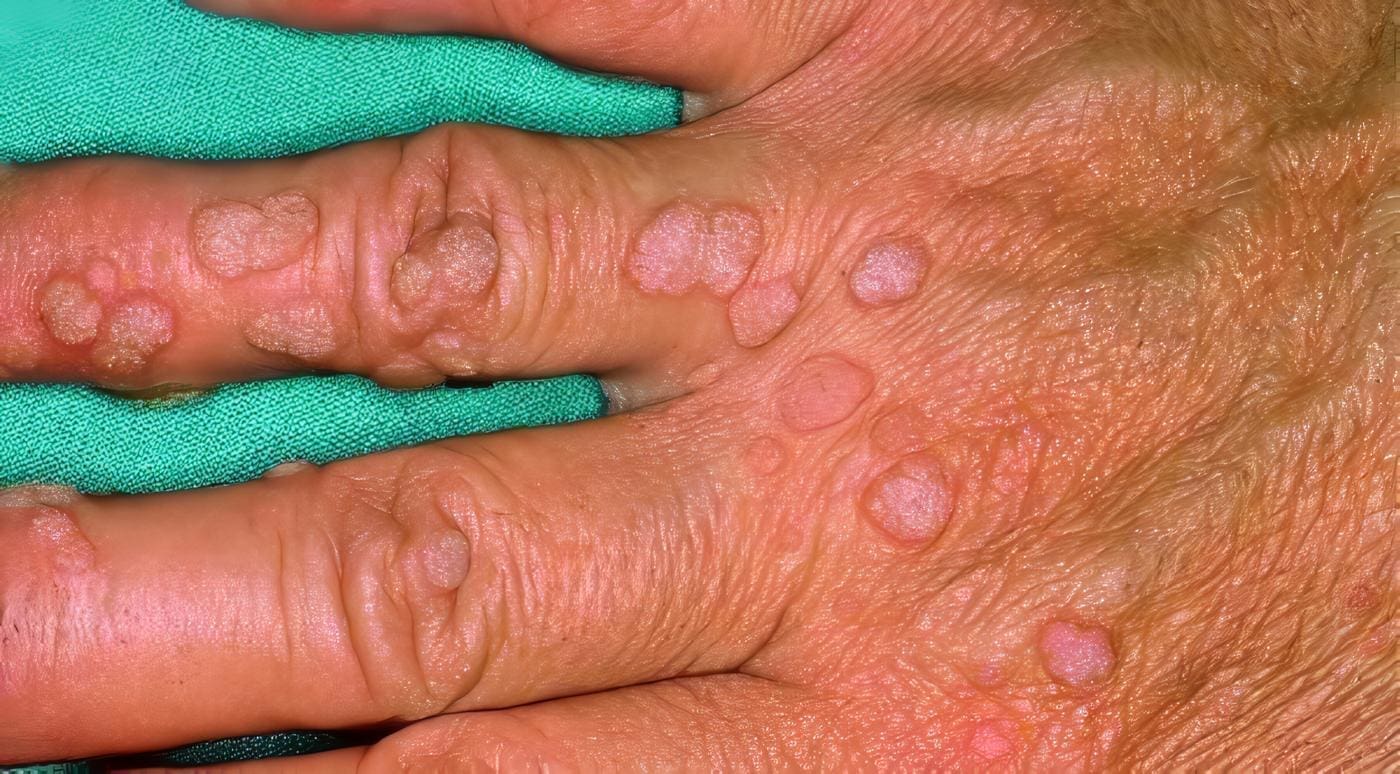
Los pacientes con SWA tienen una gran susceptibilidad a las infecciones por bacterias extracelulares y gérmenes intracelulares. Desde los seis meses de vida, cuando desaparece la IgG materna, se observan infecciones piógenas del tipo impétigo y forunculosis, y a una edad mayor, cuando se acentúa la deficiencia de los LT, se encuentran afecciones mucocutáneas por virus Herpes simplex tipo 1 y 2. Los papilomavirus producen verrugas vulgares crónicas, y el molusco contagioso es de presentación frecuente (Foto 7).
Telangiectasias mucocutáneas
Ataxia Telangiectasia (AT)

Foto 7. Síndrome de Wiskott Aldrich. Lesiones ampollosas y costrosas en ambos labios;
más acentuadas en el labio inferior. Queilitis angular (virus Herpes simplex Tipo I).
La AT es un desorden transmitido como un rasgo autosómico recesivo originado por las mutaciones que ocurren en un gen denominado ATM, el cual está localizado en el cromosoma 11q22.3; este gen codifica para una proteína importante en la regulación del ciclo celular y en la reparación del DNA (proteína ATM, quinasa localizada en los puntos de chequeo del ciclo celular), por lo anterior la AT lleva a una labilidad notable del DNA: rupturas y traslocaciones, cuando ocurre una exposición a la luz ultravioleta o a otros estímulos (20)
La enfermedad se caracteriza por deterioro neurológico progresivo, inmunodeficiencia variable, telangiectasias óculo-cutáneas, infección sinopulmonar recurrente, envejecimiento prematuro y alta predisposición al desarrollo de neoplasias.
Los defectos en la respuesta inmune son muy variables e involucran tanto la inmunidad celular como la humoral. Se encuentra atrofia tímica, linfopenia, escasa respuesta proliferativa de las células T luego del reto con mitógenos y disminución de los linfocitos T CD4+.
La hipogamaglobulinemia es frecuente, con una disminución más marcada de la IgA, la IgE y la IgG2; los LB son normales en número, pero existe un defecto en su maduración probablemente por falta de colaboración de los LT CD4+, evidenciado por una mala respuesta de anticuerpos específicos contra los antígenos protéicos y polisacáridos (20)
Las manifestaciones dermatológicas de la AT son múltiples: telangiectasias óculo-cutáneas; hirsutismo en brazos y piernas; alopecia areata; queratosis pilaris; dermatitis similar a la seborreica; acantosis nigricans, impétigo recurrente severo y verrugas vulgares múltiples (21) (Foto 8).
Dilución pigmentaria
Albinismo óculo-cutáneo

Foto 8. Ataxia Telangiectasia. Telangiectasias pronunciadas en las escleras y generalizadas en la piel de la cara.
Síndrome de Chediak-Higashi (SCH)
El SCH es un trastorno heredado como un rasgo autosómico recesivo, que se origina por mutaciones en el gen CHS1 localizado en el cromosoma 1q42-q43.
Este gen codifica para una proteína, aún en proceso de caracterización, pero se tienen evidencias de su requerimiento para la generación normal, la estructura y la función de una variedad de organelas intracelulares como los lisosomas y los gránulos secretorios (22).
Estas alteraciones llevan a la fusión anormal de los gránulos intracitoplasmáticos, originando granulaciones gigantes observadas en los leucocitos circulantes, las neuronas, los melanocitos, las células tubulares renales, los fibroblastos y el cabello.
Adicionalmente, otras consecuencias del defecto molecular son la neutropenia, por una destrucción intramedular de estos granulocitos; una disminución marcada de la quimiotaxis de los neutrófilos al parecer por un impedimento mecánico debido a los gránulos gigantes y una deficiente acción citotóxica de las células NK.