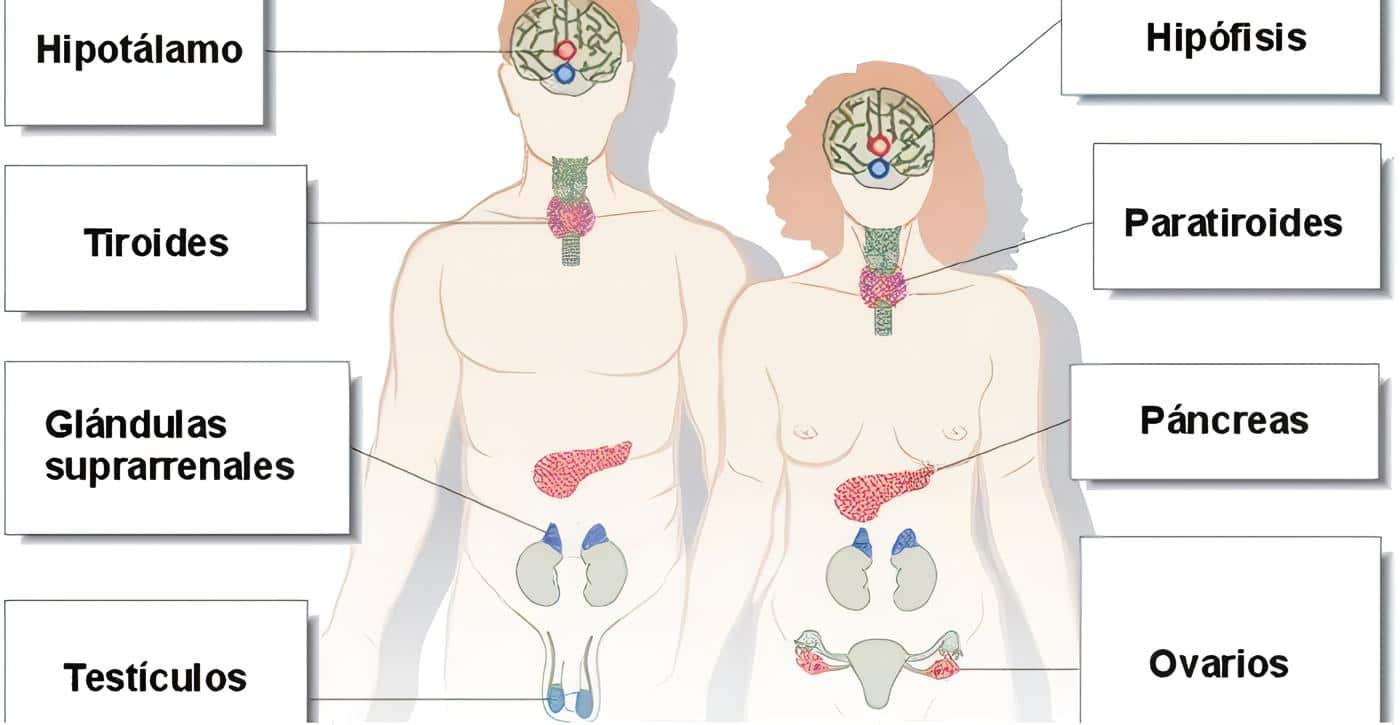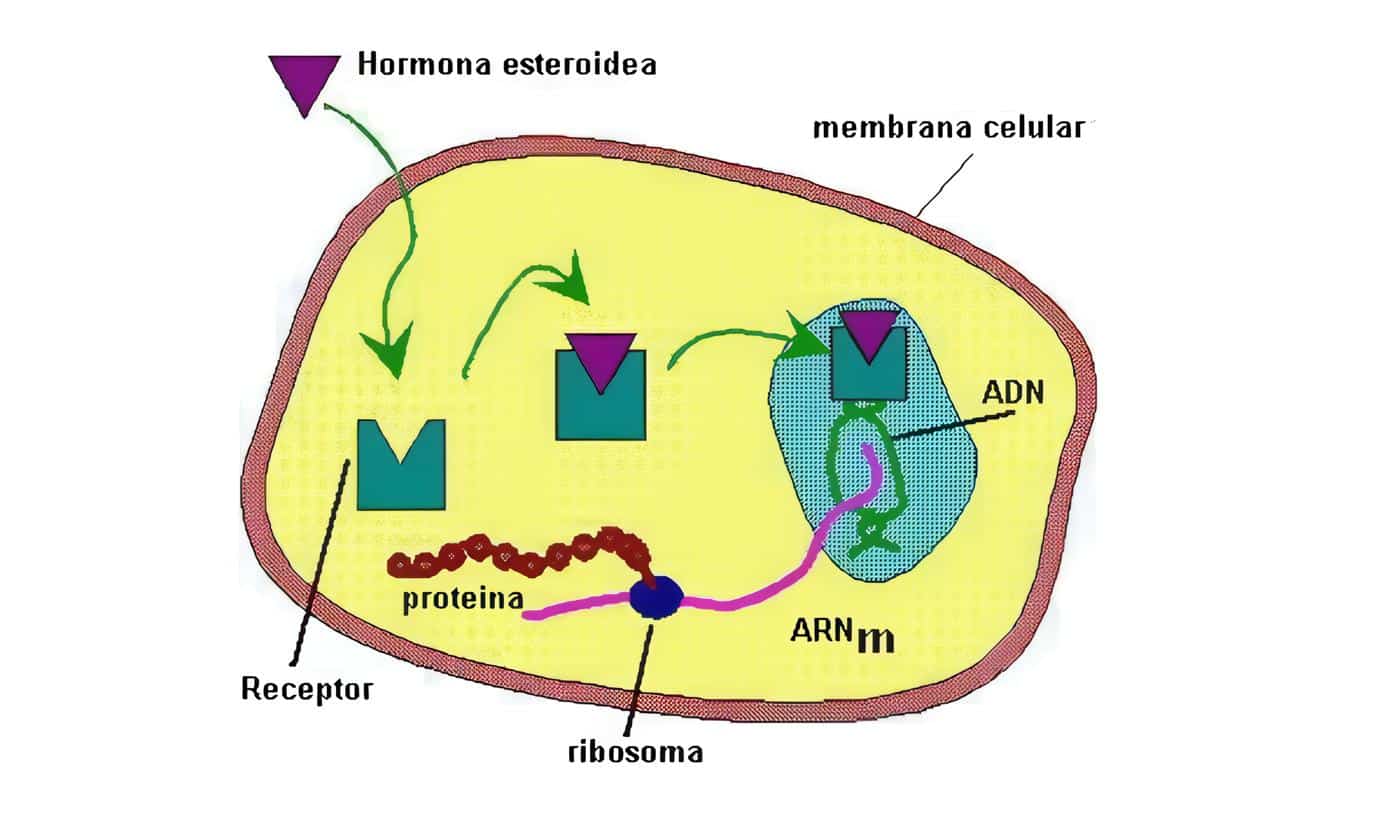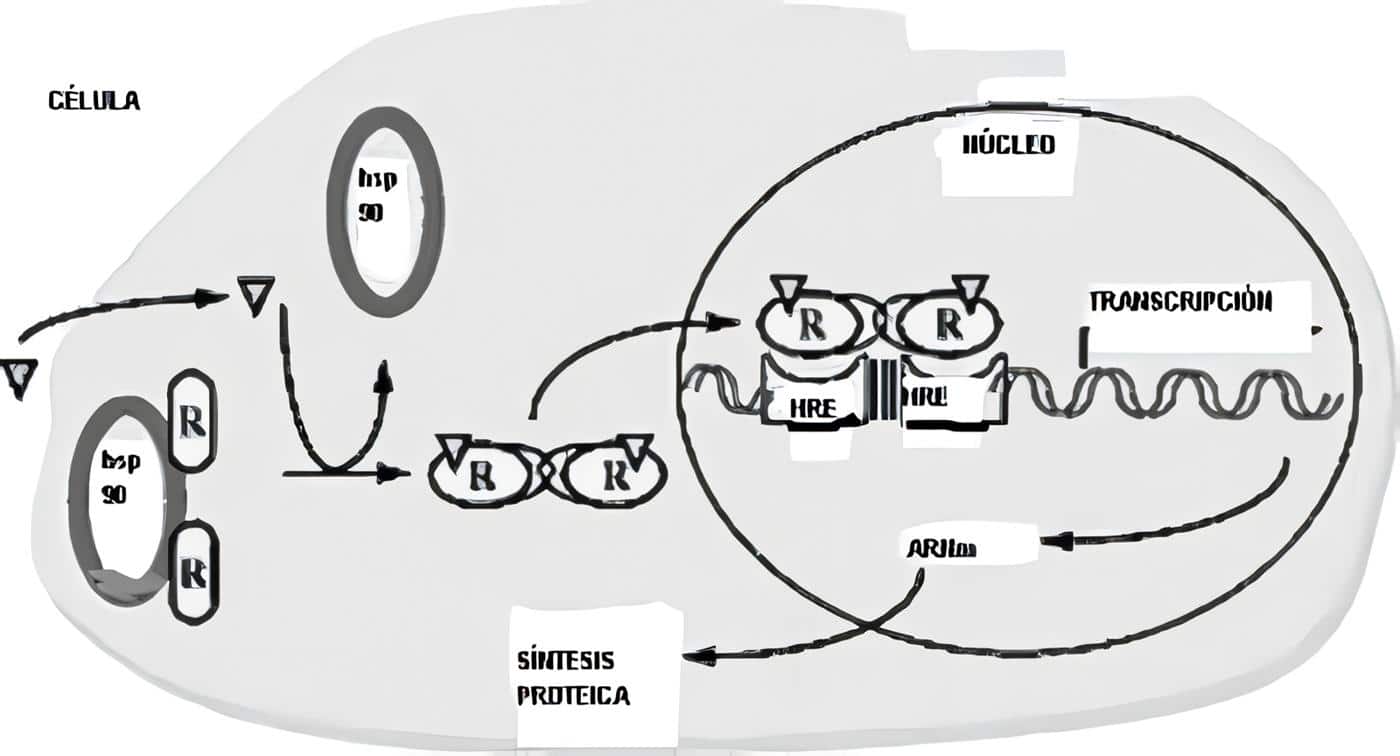
La tercera hipótesis
Del efecto de la hiperglicemia sobre el daño vascular es la activación de la proteínquinasa C. (PKC) La familia de la PKC comprende quince isoformas, nueve de las cuales son activadas por el segundo mensajero de los lípidos, el diacil glicerol (DAG).
La hiperglicemia intracelular aumenta el DAG en las células microvasculares de la retina y en el glomérulo renal de los animales diabéticos. La síntesis del DAG se aumenta a través de la reducción del fosfato de hidrooxiacetona a glicerol 3/fosfato.
Las isoformas b y d se activan primariamente a través de la hiperglicemia y también indirectamente a través de los receptores AGEs y por incremento de la vía de los polioles, aumentando a su vez las especies reactivas de oxígeno (ROS). La activación de las isoformas PCK/beta media en las anormalidades del flujo retinal y renal, posiblemente por disminución en la producción de óxido nítrico y por aumento de la actividad de la endotelina/136.
También la activación del PKC está implicada en la disminución glomerular de óxido nítrico en el músculo liso, lo cual está inducido por la hiperglicemia. La activación del PKC también inhibe la expresión del mRNA estimulada por la insulina, para la síntesis endotelial del óxido nítrico (eNOS)37. La hiperglicemia a su vez aumenta la endotelina 1 estimulada por la MAP/quinasa en el glomérulo y en el mesangio activada por las isoformas PCK.
La activación del PCK por la hiperglicemia:
También induce la expresión de la permeabilidad aumentando el VEG del músculo liso38. La hiperglicemia induce anormalidades en el flujo sanguíneo en la permeabilidad endotelial. El PKC contribuye al aumento de la matriz microvascular por inducción en la expresión del TGF/beta1, la fibrinonectina y el colágeno.
El efecto es mediado posiblemente por la inhibición en la producción del ácido nítrico por el PKC.
El PCK activado por la hiperglicemia induce la mayor expresión del factor activador inhibidor/1 del activador del plasminógeno PAI/140 El tratamiento experimental con inhibidores específicos PKC/beta reduce la actividad del PKC en la retina y en el glomérulo renal de los animales en experimentación al mejorar el tiempo de circulación, la filtración glomerular y la corrección de la excreción de la albúmina urinaria, con las mismas isoformas inhibidoras en ratones DB/DB al inhibir el PKC se mejoró la expansión acelerada del mesangio.
(Lea También: Fisiología de la Corteza Suprarrenal)
La última hipótesis es el aumento del flujo de glucosa a través de la vía de la hexosamina.
El exceso de glucosa dentro de la vía de la hexosamina causa diversas complicaciones vasculares en los animales de experimentación.
En la vía normal de la glicólisis se producen substratos que requieren UDPN/UDP N acetil glucosamina que se requiere para la síntesis del proteoglicano, con la formación de glicoproteínas unidas al oxígeno. Al inhibir esta enzima que convierte la glucosa en la glucosamina mediante una glutamino fructuosa amidotransferasa (GFAT), puede bloquearse la hiperglicemia que ha intervenido en los factores de trascripción ya mencionados TGF/alfa, TGF/beta1 y PAI/1.
Esta vía es muy importante en el papel de la hiperglicemia que induce la resistencia de la insulina, a su vez inducida por las grasas. La vía de la hexosamina y la hiperglicemia consecutiva induce también cambios en la trascripción del gen para el PAI/1.
En síntesis, la activación de la vía de la hexosamina por la hiperglicemia puede resultar en diversos cambios tanto en la expresión del gen como en la función de las moléculas proteicas que forman parte de toda la cadena de eventos que llevan a las complicaciones vasculares de la diabetes.
La hiperproducción de insulina –que lleva a síntomas adrenérgicos y neuro-glucopénicos- se presenta en el insulinoma y en la hiperplasia de las células beta, patología poco común pero que produce las mencionadas manifestaciones severas.
Terapéutica.
Brevemente diremos que el manejo de la diabetes consiste en la combinación de dieta balanceada y fraccionada, ejercicio físico y la administración de hipoglicemiantes como la insulina, o los hipoglicemiantes orales como las sulfonil-ureas, biguanidas y glitazonas. El manejo de las complicaciones requiere manejo especializado.
Historia
La diabetes –como enfermedad- se conocía desde la antigüedad. Su relación con el páncreas empezó a conocerse en el siglo XIX cuando Langerhans en 1869 describió los islotes y más tarde Minkowski produjo experimentalmente diabetes al pancreatectomizar perros.
En la búsqueda de extractos pancreáticos hipoglicemiantes – que contuvieran insulina – trabajaron Zuelzer y Gley, pero el hito lo constituyó el descubrimiento de la insulina por Banting y Best.
Los esposos Cori estudiaron la absorción y metabolismo de los azúcares, Sanger dilucidó la estructura proteica de la insulina y Steiner descubrió el precursor pro-insulina. El premio Nóbel fue otorgado a varios de estos investigadores, entre los que se encuentran Banting, Houssay, Cori, Sanger y Yalow.
El efecto diabetogènico de las hormonas contra reguladoras de la insulina fue observado por Houssay, quien notó la mejoría del perro diabético pancreatectomizado al realizar hipofisectomìas, disminuyéndose de esta forma sus requerimientos de insulina. Burger y Kramer encontraron una acción glicogenolìtica directa sobre el hígado de un preparado impuro de insulina, efecto que en realidad se debió al glucagòn.
Resumen
Los islotes pancreáticos producen insulina en sus betacélulas, glucagón en las alfa y somatostatina en las delta. Su más importante hormona es la primera, que a través de estímulos alimenticios como la glucosa, proteínas y cuerpos cetónicos- regulan fundamentalmente la utilización y homeostasis de la glucosa.
Por tratarse de una proteína de cincuenta y un aminoácidos –acomodados en dos cadenas ligadas por puentes disulfídicos- circula en forma libre, como un polímero de tres monómeros.
Los tejidos que principalmente responden a la insulina a través de receptores en sus membranas son el muscular y el adiposo, pues el hígado y el sistema nervioso son permeables a la glucosa, aunque la insulina produce una serie de enzimas hepáticas y de otros tejidos, favoreciendo la formación del glicógeno y la glicólisis.
La insulina (una hormona anabólica) es contra-regulada por las hormonas del estrés –glucagón, epinefrina, cortisol (con efectos catabólicos) y la hormona del crecimiento (con efectos mixtos)- que regulan la homeostasis de la glucosa.
Un déficit absoluto o relativo de la insulina se encuentra involucrado en el desarrollo de la diabetes mellitus, el trastorno metabólico más frecuente y grave que se conoce.
Referencias seleccionadas
- Adrian TE el al. Secretion of pancreatic polypeptide in patients with pancreatic endocrine tumors. N Eng J Med 1986; 315: 287-291.
- Kieffer TJ, Habener JF. The glucagons-like peptides. Endocrine Reviews 20 (6): 876-913.
Kroetz DL. Nuclear receptors, how do they regulate expression?
https://www.aapspharmaceutica.com/inside/focus_groups/drugtrans/imagespdfs/kroetz.pdf - Evans JL, Goldfine B, Maddux A, Grodsky GM. Oxidative Stress and Stress-Activated Signaling Pathways: A Unifying Hypothesis of Type 2 Diabetes. Endocrine Reviews 2002; 23 (5): 599-622
- Sánchez-Medina M. Integración de un paradigma, mecanismos biomoleculares y patogénicos precursores de las complicaciones de la diabetes. Presentación en la Academia Nacional de Medicina de Colombia, Junio 24 de 2004.
- Watson RT, Kankasi M, Pessin JF. Regulated Membrane Trafficking of the Insulin-Responsive Glucose Transporter 4 in Adipocytes. Endocrine Reviews 2004; 25 (2): 177-204.
- Watson RT, Pessin JE. Intracellular Organization of Insulin Signaling and GLUT4 Translocation. Recent Progress in Hormone Research 2001; 56:175-194 (2001)
- Schmitz O et al. Amylin agonists, a novel approach in the treatment of diabetes. Diabetes 2004; 53 (S3): S233-8.
- Baggio LL, Drucker DJ. Harnessing the therapeutic potential of glucagon-like peptide-1, a critical review. Treat Endocrinol 2002; 1:117-25