Alteraciones en el Tamaño Celular
Las anemias microcíticas y macrocíticas, aunque no son características de un fenotipo particular de IDP, se observan frecuentemente y pueden relacionarse con el estado fisiopatológico del paciente.
Estas anemias en los pacientes con IDP pueden ser secundarias a la deficiencia de vitamina B12 o ácido fólico (hipoplasia cartílago-pelo, IDCV), pueden ser consecuencia de procesos hemolíticos (síndrome de Hiper-IgM), pérdidas sanguíneas (síndrome de Wiskott-Aldrich) y, en la mayoría de los casos se deben a procesos infecciosos e inflamatorios crónicos (1, 13).
La presencia de blastos es otro hallazgo común en el ESP de los pacientes con IDP, especialmente en aquellas enfermedades relacionadas con defectos en la maduración y producción celular en la médula ósea (13).
Alteraciones en los Gránulos Citoplasmáticos
Granulaciones o Vacuolaciones Tóxicas
Es una alteración morfológica de los PMN caracterizada por la presencia de granulaciones azurófilas pesadas, vacuolaciones citoplasmáticas y cuerpos de Dolhe. Estos “neutrófilos tóxicos” no son específicos de una condición en especial, pero se observan principalmente en individuos con enfermedades que cursen con inflamaciones agudas y crónicas, tales como las infecciones bacterianas severas.
Debido a que estas infecciones son comúnmente observadas en los pacientes con IDP, es factible la presencia de granulaciones tóxicas en los PMN de estos individuos (13).
Síndrome de Chediak-Higashi:
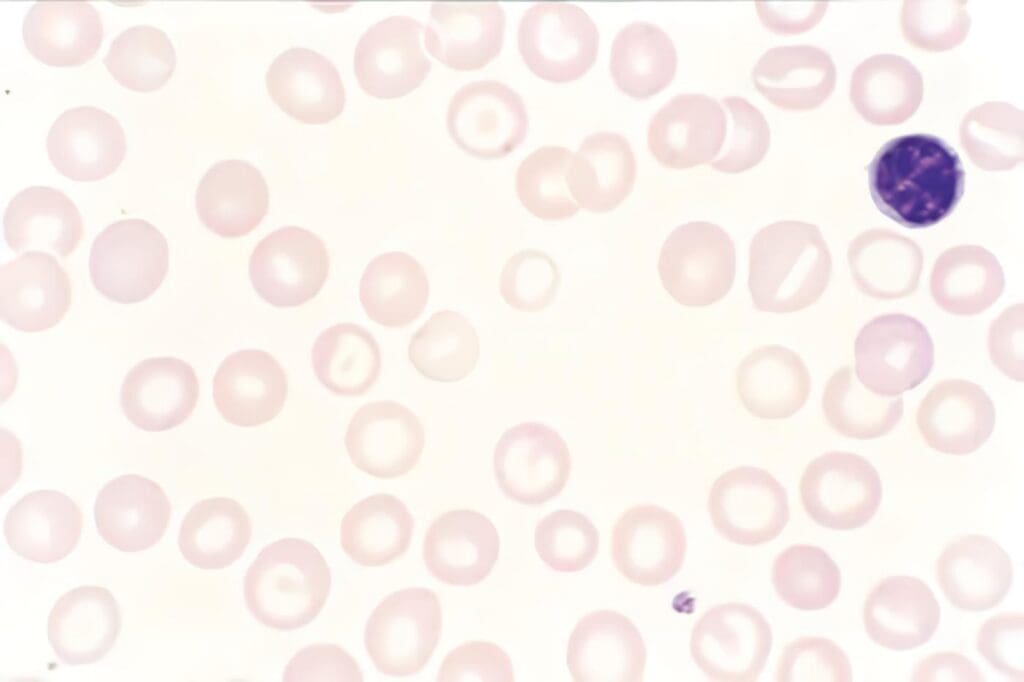
Es un desorden genético que se origina por mutaciones en el gen LYST, el cual codifica para una proteína importante en la función del citoesqueleto. En esta IDP, el ESP se caracteriza por la presencia de lisosomas gigantes en los PMN y otros leucocitos (Figura 1).
Estos cuerpos, que en los PMN se generan por la fusión de los gránulos peroxidasa positivos, son fácilmente demostrables por la microscopía de rutina, elemento esencial en el diagnóstico de esta enfermedad. Además, en esta IDP se presenta neutropenia, trombocitopenia, pérdida de la función de las células asesinas naturales y, en etapas avanzadas, se puede observar pancitopenia (3, 4, 13, 24).
Conclusiones
El ESP y los datos obtenidos a partir de él deben ser mejor explotados en la práctica clínica. Este examen básico, de fácil realización y bajo costo, está al alcance de todos los laboratorios clínicos y, si existe buena calidad en su preparación y análisis, permite la búsqueda, hallazgo y seguimiento de diferentes enfermedades.
El ESP es una herramienta de gran valor en la evaluación del paciente con infección recurrente, pues sus hallazgos cualitativos y cuantitativos son claves para la orientación diagnóstica inicial de una IDP. Aunque esta prueba requiere de compromiso y dedicación por parte del laboratorista clínico, su uso permite realizar mejores diagnósticos en el laboratorio y por tanto una mejor orientación clínica, aspecto vital en el caso de los pacientes con IDP.
Debido a que las pruebas específicas para el diagnóstico de las IDP son costosas, toman un tiempo importante, no se realizan de rutina en los laboratorios clínicos y existen pocas personas capacitadas para su ejecución, se debe rescatar la importancia del desarrollo de las pruebas no específicas, en especial del ESP, como herramienta fundamental para seleccionar los pacientes que requieran ESTUDIOS más avanzados.
Referencias Bibliográficas
- 1. Nathan D, Orkin S. Nathan and Oski´s Hematology of infancy and childhood. En: Grupp S, Abbas A. Humoral Immunity and the Development and Regulation of Immune Responses. 5th ed. Toronto: W.B. Saunders Company, 1998:1023-1050.
- 2. García de OD, Patiño PJ, Salgado H, Montoya CJ, López JA, Pérez JE. Evaluación del paciente con inmunodeficiencia. Síndrome de infección recurrente patológica. Medicina y Laboratorio 1997, 7:545-575.
- 3. Holland S, Gallin J. Evaluation of the patient with recurrent bacterial infections. Ann Rev Med 1998, 49:185-199.
- 4. IUIS Scientific Committee. Primary immunodeficiency diseases. Report of an IUIS scientific committee. Clin Exp Immunol 1999, 118 (Suppl. 1):1-28.
- 5. Carneiro-Sampaio M, Grumach A, Manissadjian A. Laboratory screening for the diagnosis of children with primary immunodeficiencies. J Invest Allergol Clin Immunol 1991, 1:95-200.
- 6. Lee R, Foerster J, Lukens J, Paraskevas F, Greer P, Rodgers G. Wintrobe’s Clinical Hematology. 10th ed. Philadelphia: Lippincott Williams & Wilkins, 1998:1484.
- 7. Houwen B. Blood Film Preparation and staining procedures. Lab Hematol 2000, 6:1-7.
- 8. Vélez H, Rojas W, Borrero J, Restrepo J. Fundamentos de Medicina. Hematología. En: Restrepo A. El Leucograma, Mielofibrosis, Mononucleosis Infecciosa. 5th ed. Medellín: CIB, 1998:126-135.
- 9. Campuzano G. Semiología del hemograma. Laboratorio al Día 1995, 5:161-174.
- 10. Beutler E, Coller B, Lichtman M, Kipps T, Seligsohn U. William’s Hematology. 6th ed. New York: MacGraw-Hill, 2001:1941.
- 11. Behrman R, Kliegman R, Jenson H. Nelson. Tratado de Pediatría. En: Buckley R. El sistema inmunitario y sus transtornos. 16th ed. Barcelona: McGraw-Hill, Interamericana, 2000:645-706.
- 12. Campuzano G. ESTUDIO del paciente con neutropenia. Laboratorio Al Día 1996, 6:11-28.
Fuentes Bibliográficas
- 13. Smith C, Miller D, Baehner R. Blood diseases of infancy and childhood. 17th ed. New York: Mosby, 1995:1041.
- 14. Ochs HD, Smith C, Puck J. Primary Immunodeficiency Diseases. A molecular and genetic approach. New York: Oxford University Press, 1999:501.
- 15. Ginzberg H, Shin J, Ellis L. Schwachman syndrome: phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar. J Pedriatr 1999, 135:81-88.
- 16. Aprikyan A, Liles W, Park J. Myelokathexis: a congenital disorder of severe neutropenia characterized by accelerated apoptosis and defective expression of bcl-x in neutrophil precursors. Blood 2000, 135:320-327.
- 17. Auten K, Frank M, Atkinson J. Samter´s Immunologic Diseases. En: Holland S, Gallin J. Phagocyte Disorders: Lippincott Williams & Wilkins, 2001:329-349.
- 18. Markert L, Hummell D, Rosenblat H. Complete DiGeorge syndrome: persistence of profound immunodeficiency. J. Pediatr 1998, 132:15-21.
- 19. Ridanpää M, Eenennam H, Pelin K, Chadwick R, Johnson C, Yuan B. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, Cartilage-Hair Hypoplasia. Cell 2001, 104:195-203.
- 20. Snapper S, Rosen F. The Wiskott-Aldrich syndrome protein (WASP): roles in signaling and cytoskeletal organization. Ann Rev Immunol 1999, 17:905-925.
- 21. Remold-O’Donnell E, Rosen F, Kenney D. Defects in Wiskott-Aldrich syndrome blood cells. Blood 1996, 87:2621-2631.
- 22. Shcherbina A, Rosen F, Remold-O’Donnell E. WASP levels in platelets and lymphocytes of Wiskott-Aldrich syndrome patients correlate with cell dysfunction. J Immunol 1999, 163:6314-6320.
- 23. Campuzano G. Estudio del paciente con trombocitopenia. Laboratorio al Día 1995, 5:25-350.
- 24. Brostoff J, Gray A, Male D, Roitt I. Case Studies in Immunology. Companion to Inmmunology. En: Immunodeficiency. 5th ed. Philadelphia: Mosby, 1998:13 – 30.