
Terapia génica
Según la Sociedad Americana de Terapia Génica y Celular, la terapia génica se puede definir como el conjunto de estrategias que modifican la expresión de un gen o que corrigen genes anormales, mediante la administración de un ácido nucleico (ADN o ARN) específico (69).
Las MPS son excelentes candidatas para su tratamiento por terapia génica por las siguientes razones: (i) teóricamente solo el 10% de la actividad enzimática permite pasar de un fenotipo severo a uno atenuado, pues valores cercanos a este porcentaje, e incluso inferiores, son encontrados en pacientes con las variantes medias y atenuadas de la enfermedad (70), (ii) no es necesaria una regulación estricta del transgen, debido a que los genes de las enzimas lisosomales presentan expresión constitutiva (56), (iii) es posible realizar la corrección del defecto en células no transducidas por el vector gracias al mecanismo de corrección cruzada mediada por el receptor de manosa-6-fosfato (71), (iv) ensayos preclínicos en diferentes modelos animales (ratón, rata, perro ygato) han permitido niveles enzimáticos terapéuticos hasta por 1,5 años en ratón (72) y 3 años en perros (73), con importantes correcciones bioquímicas y clínicas, y (v) en las enfermedades producidas por la deficiencia de una sulfatasa, la coexpresión conSUMF1 ha permitido aumentos significativos en la actividad de sulfatasa recombinante (74, 75). Diferentes vectores han sido empleados para realizar la transferencia génica en modelos animales de EDL, siendo los más frecuentes los lentivirus, adenovirus y virus adenoasociados (13, 76, 77).
El primer trabajo de transferencia génica para la MPS IVA fue el desarrollado por Toietta et al. (17), en el que se empleó un vector retroviral portando el gen de GALNS para transducir fibroblastos y linfocitos de sangre periférica de individuos normales y pacientes Morquio A, así como queratinocitos humanos, mioblastos de ratón y sinoviocitos de conejo. El uso de este vector permitió obtener valores de actividad enzimática entre 5 y 50 veces por encima de los encontrados en células normales, así como una reducción de los GAGs a niveles iguales o menores a los encontrados en células normales.
Durante los últimos años el Instituto de Errores Innatos del Metabolismo en colaboración con la Universidad de Saint Louis (USA), ha trabajado en el desarrollo de una estrategia de terapia génica para el tratamiento de la enfermedad de Morquio A. Para este objetivo se seleccionaron vectores derivados de virus adenoasociados (AAV). Los AAV son virus no envueltos pertenecientes a la familia Parvoviridae genero Dependovirus, constituidos por un genoma de ADN de cadena sencilla de 4,7 kb (78). Dentro de los diferentes vectores empleados en terapia génica, los vectores derivados de AAV han ganado importancia durante los últimos años por presentar un buen perfil de seguridad, no estar asociados con la generación de fuertes efectos adversos, y bajo ciertas condiciones, permitir la expresión del transgen por periodos prolongados de tiempo (78-80). En el caso de los MPS estos vectores han sido empleados en modelos animales de MPS I, II, IIIA, IIIB, VI y VII (13, 76).
Los primeros trabajos para el uso de vectores AAV en MPS IV A estuvieron encaminados al establecimiento y apropiación de las técnicas de clonación y producción de los vectores. En esta etapa se demostró el aumento de la actividad enzimática en fibroblastos MPS IV A y células HEK293 transducidas con los vectores AAV. Este estudio constituyó el primer trabajo de transferencia génica utilizando vectores virales en el país, y mostró la posibilidad de realizar la corrección del defecto genético en pacientes con MPS IV A empleando vectores AAV (16). Sin embargo, por esa época algunas publicaciones comenzaron a mostrar las limitaciones del uso de promotores virales para realizar la expresión del gen de interés (81). Debido a que el vector previamente construido empleaba el promotor del citomegalovirus (CMV), en el siguiente trabajo se enfocó en el estudio de promotores no virales que permitieran obtener niveles de expresión similares al obtenido con el promotor CMV, y a la evaluación de los mismos en cultivos celulares y en el modelo murino de la enfermedad.
La primera parte del trabajo comparó in vitro el efecto de promotor del CMV con los promotores eucarióticos del factor de elongación 1a (EF1) y de la a1 antitripsina humana (AAT) (14). Los resultados mostraron que los promotores eucarióticos (AAT y EF1) permitian niveles enzimáticos similares a los obtenidos con el promotor CMV, sin observar una regulación negativa del promotor CMV, lo cual difería de los reportes de silenciamiento de este promotor observados por nosotros y otros grupos de investigación (15, 81). Sin embargo, esos estudios empleaban vectores retrovirales, adenovirales o plásmidos (82-85), y hasta ese momento no se había realizado un estudio detallado del silenciamiento del promotor CMV en el contexto de un vector AAV2. De esta manera los resultados de esta primera etapa sugirieron, por primera vez, la existencia de un mecanismo inducido por el vector AAV que evitara el silenciamiento del promotor CMV, lo que podría explicar los reportes en los que el uso del promotor CMV y un vector AAV han permitido la expresión de diferentes genes por periodos hasta de un año y medio en ratones (81, 84, 86, 87).
Otro de los factores importante en el desarrollo de una terapia génica para la enfermedad de Morquio A es la coexpresión con SUMF1. En este aspecto se logró demostrar que la coexpresión con SUMF1, independiente del promotor utilizado, permitía un aumento hasta de 4 veces en la actividad enzimática en el lisado celular (14). Por otro lado, la actividad en el medio de cultivo fue detectada exclusivamente en células coexpresando GALNS y SUMF1, mostrando la importancia de la coexpresión con SUMF1 para lograr que la enzima producida sea exportada y pueda ser capturada por las células no transducidas. Un hallazgo importante, y no reportado hasta ese momento, fue la variación del efecto de la coexpresión con SUMF1 observada en diferentes tipos celulares. Mientras en células HEK293 la actividad en el medio de cultivo era detectada con relaciones GALNS: SUMF1 1:1, en fibroblastos era necesario utilizar relaciones GALNS: SUMF1 1:2 para lograr detectar la enzima en el medio de cultivo; mientras que en condrocitos murinos el aumento fue menor que el observado en células HEK293 y fibroblastos. En resumen, estos resultados mostraron la importancia de la coexpresión con SUMF1 y de la selección adecuada de la relación GALNS: SUMF1 dependiendo del tipo de células transducidas, para lograr niveles terapéuticos de actividad enzimática.
En la siguiente etapa se realizó la evaluación in-vivo de la terapia mediante la administración del vector AAT-GALNS, o la coadministración de AATGALNS y CMV-SUMF1, en ratones MPS IV A (Tabla 2) (88). Al cabo de 12 semanas, la administración del vector AAT-GALNS permitió niveles de actividad enzimática en plasma del 8%, los cuales llegaron a ser cercanos al 20% en animales coinyectados con los vectores AAT-GALNS y CMV-SUMF1. En tejidos los mayores niveles se observaron en hígado, con un 21 y 37% de los valores normales, para animales AAT-GALNS y AAT-GALNS: CMVSUMF1, respectivamente. En este último grupo de animales, se destacó el aumento significativo de la actividad enzimática en corazón y hueso (30 y 33% de los valores normales, respectivamente), dos de los tejidos más afectados tanto en el modelo murino como en los pacientes con MPS IVA, mostrando la ventaja de la coadministración con SUMF1. Otro hallazgo importante fue el hecho de que no se observó silenciamiento del promotor CMV durante las 12 semanas del estudio, ratificando lo observado en los ensayos in vitro y sugiriendo nuevamente que el silenciamiento de este promotor puede estar estrechamente ligado al tipo de vector utilizado. Este trabajo constituyó la primer evidencia in vivo de la posibilidad de realizar la corrección del defecto genético en la enfermedad de Morquio A (88).
Durante el desarrollo de las etapas anteriores se observó que los vectores AAV no poseían afinidad por tejido óseo, limitando así la entrega del gen de interés a este tejido. El hueso es un tejido de soporte vascularizado consistente en células y una matriz extracelular mineralizada. Cerca de 100 millones de personas sufren a nivel mundial de enfermedades relacionadas con problemas óseos, incluyendo osteoporosis, cáncer, osteoartritis, artritis inflamatoria y algunos errores innatos del metabolismo (89). Por lo tanto, un agente terapéutico diseñado para el tratamiento de estas enfermedades debe ejercer principalmente su actividad farmacológica en este tejido. La hidroxiapatita es el principal componente inorgánico del hueso y se encuentra presente de forma exclusiva en este tejido, por lo que constituye un blanco importante para el direccionamiento de agentes terapéuticos a tejido óseo (90). Tomando como base la estrategia de direccionamiento a hueso empleada para la enzima GALNS recombinante (11), la tercera fase del proyecto consistió en adaptar esta tecnología para el direccionamiento a hueso de un vector AAV. Para este fin, se construyó un vector AAV portando la señal de direccionamiento a hueso en su cápside, con el objetivo de alterar el tropismo natural del vector y permitir su biodistribución a hueso (Figura 5).
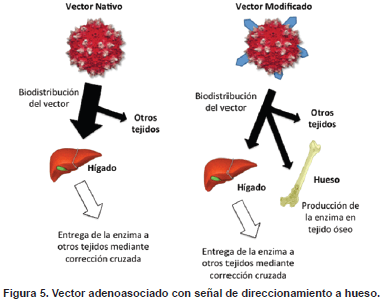
La señal de direccionamiento a hueso fue insertada en la cápside del vector adenoasociado nativo para aumentar la biodistribución del vector a hueso. Mientras con el vector nativo la enzima llega a tejidos no transfectados (incluyendo hueso) mediante el mecanismo de corrección cruzada, con el vector modificado las células óseas son capaces de producir su propia enzima.
La cápside de los AAV está formada por las proteínas VP1, VP2 y VP3 (Figura 6a) que se encuentran en una relación 1:1:10. Estudios de mutagénesis han mostrado que existen diferentes posiciones dentro de las proteínas de la cápside en las que se puede realizar la inserción de péptidos sin alterar significativamente los procesos de empaquetamiento y transducción (91, 92). Dentro de estas posiciones, el extremo N-terminal de la proteína VP2 representa uno de los sitios más estudiados debido a que no se afecta el sitio de unión utilizado por el virus para entrar a la célula (93). A pesar que un gran número de estudios han mostrado los beneficios de realizar la modificación de la cápside del vector para aumentar su afinidad por células pulmonares, endoteliales, islas pancreáticas, tejido vascular, músculo, miocardio o células cancerígenas (93), hasta el momento no se había evaluado el direccionamiento de un vector AAV a hueso.

El genoma de los vectores AAV esta conformado por el gen Rep, que codifica para las proteínas Rep encargadas de la regulación de los procesos de replicación, empaquetamiento e integración, y el gen Cap que codifica para las proteínas de la cápside VP1, VP2 y VP3, cuya diferencia radica en el codón de inicio empleado. La secuencia de ADN codificando para la señal de direccionamiento a hueso fue insertada inmediatamente después del codón de inicio de VP2, dentro del plásmido de empaquetamiento.
La inserción de la señal de direccionamiento a hueso se realizó en el extremo N-terminal de la proteína VP2 (Figura 6b) mediante mutagénesis sitio dirigida en el plásmido de empaquetamiento. Luego de comprobar la correcta inserción de la secuencia codificando para la señal de direccionamiento, el vector fue evaluado tanto in-vitro como in-vivo para comprobar su capacidad de empaquetaminto, transducción, afinidad por hidroxiapatita, biodistribución y expresión. In-vitro, el vector modificado mostró una afinidad del 100% por la hidroxiapatita, a diferencia de lo observado con el vector nativo, el cual presentó un porcentaje de afinidad inferior al 1%. Los estudios en cultivos celulares mostraron que la presencia de la señal de direccionamiento no afectó la capacidad de transducción del vector, mientras que en los ensayos in-vivo el vector con la señal de direccionamiento a hueso permitió detectar el transgen en concentraciones hasta 300 veces más elevadas que las observadas con el vector nativo. In-vivo el vector modificado fue capaz de liberarse de la hidroxiapatita y transducir las células adyacentes permitiendo valores de actividad enzimática en hígado, corazón, cerebro, médula ósea y hueso, mayores que los observados en animales inyectados con el vector sin modificar (Tabla 2) [(94) y datos sin publicar]. Estos resultados muestran el potencial de este vector modificado como herramienta importante no solo para el tratamiento de la MPS IV A, sino también para el tratamiento de otras enfermedades con problemas óseos como artritis, hipofosfatasia y errores innatos del metabolismo con compromiso óseo.





Creo que debería de haber mas solidaridad con estos pacientes necesitan que les brinden mejor calidad para que ellos puedan estar en mejores condiciones de salud ..ponernos en los zapatos de cada personita con este síndrome que es bastante devastador…y sugiere muchos cuidados y un buen tratamiento para que se sienten mucho mejor y puedan ser más independientes y realizar algunas metas que sean puesto ..ya que tengo una hija con este síndrome y le hecho mucha mente a esto me preocupa bastante por que quiero que mi hija esté en mejores condiciones de vida la amo y quiero verla bien e independientemente muchas gracias
Excelente documento , tengo un hijo de 4 años al cual le diagnosticaron morquio IV A . Está en tratamiento con reemplazo enzimatico cada 8 días.Segun el documento quiero preguntar si se han hecho prueba en humanos y si no para cuando lo harían ya que quiero ayuda a mi hijo para que tenga mejor calidad de vida Y tratar de contactarme con alguien a niveles superiores de nuestra medicina interna
Agradezco su respuesta
Mario buenas tardes, encolombia es un portal de contenido. Este artículo hace parte de una publicación de la academian Nacional de Medicina, debes consultar con ellos directamente.