Un vector de clonación, es un sistema genético replicante cuya replicación es autónoma e independiente de la de otros sistemas coexistentes. Así pues, la definición de vector restringe a ciertas moléculas de DNA como plásmidos, virus o derivados de ellos, la gama de vectores que se emplean actualmente para los experimentos de DNA recombinante. Como cualquier sistema replicante, un vector debe poseer un origen de replicación que le permita su duplicación autónoma del DNA cromosómico de la célula que lo aloja y también debe contener marcas genéticas que permiten la selección de la célula que los contiene.
Tabla 1. Nombre, denominación taxonómica y secuencias de nucleótidos reconocidas por algunas endonucleasas de restricción del tipo II empleadas en experimentos de DNA recombinante.
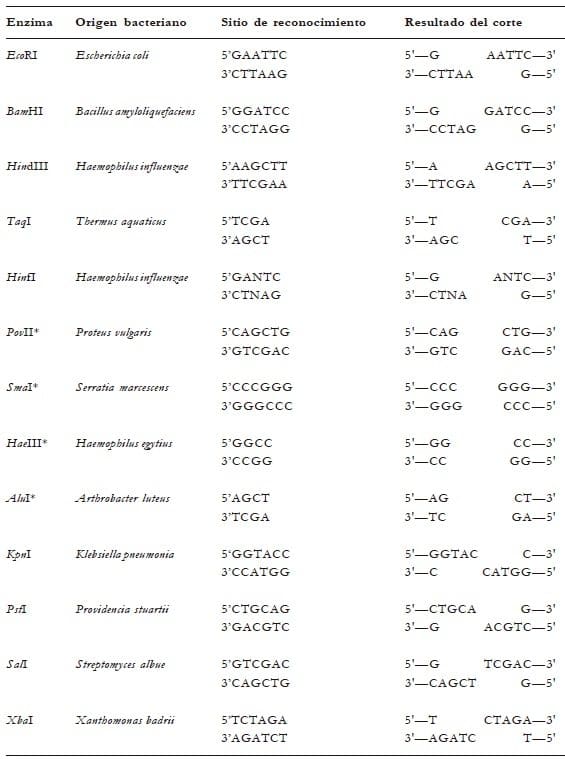
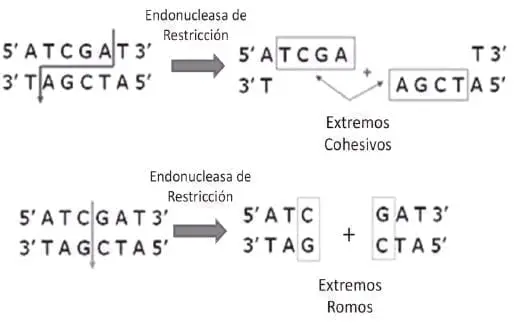 Figura 4. Enzimas de restricción. Este tipo de endonucleasas puede generar dos tipos de extremos. En el primer caso, el corte genera nucléotidos de cadena única protuberantes llamados extremos cohesivos. Estos extremos se pueden unir por medio de otra enzima, la DNA ligasa. En el segundo caso, se generan extremos doble cadena (extremos romos). Estos extremos también pueden ser unidos con la ayuda de una enzima ligasa en condiciones fisicoquímicas especiales.
Figura 4. Enzimas de restricción. Este tipo de endonucleasas puede generar dos tipos de extremos. En el primer caso, el corte genera nucléotidos de cadena única protuberantes llamados extremos cohesivos. Estos extremos se pueden unir por medio de otra enzima, la DNA ligasa. En el segundo caso, se generan extremos doble cadena (extremos romos). Estos extremos también pueden ser unidos con la ayuda de una enzima ligasa en condiciones fisicoquímicas especiales.
La clonación de un fragmento de DNA en un vector permite producir cantidades DNA recombinante a partir de una molécula original. Se define un clon como una población de moléculas o células todas idénticas con relación a una molécula o célula ancestral original. Por esta razón a la técnica de DNA recombinante también se le denomina clonación molecular. La clonación de DNA se puede realizar gracias a la capacidad que poseen los plásmidos bacterianos y fagos para continuar sus «estilos de vida usuales» una vez que se les han insertado secuencias de DNA foráneo a sus genomas. Una inserción de DNA genera un vector híbrido o quimérico que consiste en parte de DNA del genoma original y una parte de secuencias adicionales o «foráneas». Estos elementos quiméricos replican en bacterias de forma similar a los vectores no recombinantes, de esta forma se pueden obtener grandes cantidades de un fragmento foráneo clonado en estos.
Para clonar DNA existen una serie de vectores, los cuales han sido objeto de manipulación, que incluyen una gama muy amplia de plásmidos bacterianos derivados y modificados de contrapartes naturales, bacteriófagos dentro de los que se incluye una amplia de modificados del bacteriófago Lambda, del M13 y de otros. Además una serie muy importante de vectores de clonación contienen elementos genéticos derivados de sistemas eucarióticos, que permiten la manipulación y expresión de genes bajo condiciones de expresión eucarióticas. Finalmente se han desarrollado vectores derivados de virus de animales y plantas, los cuales se emplean para producir organismos transgénicos y en la terapia génica que revisaremos más adelante.
Los plásmidos, son quizá los vectores más utilizados. Estos son moléculas circulares de DNA que se replican de manera independiente al cromosoma de la célula hospedera. De manera natural se encuentran en las bacterias en tamaños que van desde 5,000 hasta 400,000 pb. Existen dos tipos generales de sistemas de control de replicación del plásmido: aquellos de copia única son mantenidos a niveles de 1 plásmido por bacteria en contraste con los multicopia que se encuentran en niveles mucho mayores, típicamente de 10 a 20 genomas por célula.
El plásmido pBR322 es uno de los más célebres que existen, por ser el primer plásmido diseñado y utilizado en biotecnología. Aunque se basa en un andamiaje natural, le fueron agregados genes para realizar las funciones que el investigador requería. Construido por Francisco Bolívar y Raymond Rodríguez de la Universidad de Stanford, se convirtió en una de las primeras herramientas para la introducción de genes específicos en una bacteria. El pBR322cuyo tamaño es de 4361 pares de bases, posee un gen de resistencia a la ampicilina que le confiere a la célula portadora del plásmido la capacidad de crecer en un medio bacteriológico con dicho antibiótico, esta resistencia se destruye cuando este se somete a tratamiento con EcoRI. Es así que si un DNA es clonado en este sitio, el recombinante es sensible al tratamiento con este antibiótico pero conserva la resistencia a tetraciclina (figura 5). Este doble juego permite establecer un sistema muy rápido y sencillo de seleccionar clones recombinantes cuyos fragmentos de DNA clonados han sido ligados en este sitio. Este procedimiento se denomina «selección negativa», porque se busca que la colonia de bacterias o levadura pierda una función; en este caso, la de resistir a la ampicilina.
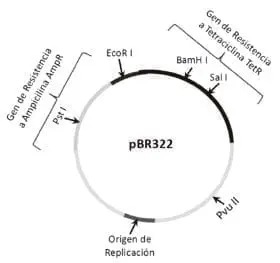 Figura 5. Mapa genético y de restricción del plásmido vector pBR322. Como vector de clonación este plásmido desarrollado por Francisco Bolívar y Raymond Rodríguez contiene un solo sitio para el corte con EcoRI que separa el gen de resistencia a Ampicilina AmpR y BamHI cuya acción inactiva el gen de resistencia a tetraciclina TetR. Además contiene un sitio de origen de replicación OriC que le permite una replicación autónoma. El tamaño total del plásmido es de 4361 pb siendo su capacidad de clonación de aproximadamente 10.000 pares de bases.
Figura 5. Mapa genético y de restricción del plásmido vector pBR322. Como vector de clonación este plásmido desarrollado por Francisco Bolívar y Raymond Rodríguez contiene un solo sitio para el corte con EcoRI que separa el gen de resistencia a Ampicilina AmpR y BamHI cuya acción inactiva el gen de resistencia a tetraciclina TetR. Además contiene un sitio de origen de replicación OriC que le permite una replicación autónoma. El tamaño total del plásmido es de 4361 pb siendo su capacidad de clonación de aproximadamente 10.000 pares de bases.
Se han construido muchos plásmidos para ser usados como vectores de clonación. La gran mayoría de ellos reúnen varias características genéticas bacterianas o de sistemas virales eucarióticos, estas permiten desarrollar estrategias de tamizado de secuencias específicas clonadas.
Como se ha descrito antes, talvez, la estrategia más común es el uso de plásmidos vectores que portan genes que especifican por resistencia a antibióticos. Esta característica fue exitosa en el diseño de un sistema de clonación. Sin embargo, no es la única manera de tener un sistema de tamizado. A partir de la introducción de genes que codifican por enzimas cuya actividad puede ser fácilmente monitoreada, se han construido una serie de vectores que expresan la beta-Galactosidasa. Esta enzima hidroliza la lactosa en sus dos monosacáridos glucosa y galactosa. La actividad enzimática puede ser trazada utilizando un sustrato artificial acoplado a una molécula que da una reacción coloreada (azul). Si el DNA foráneo es clonado cerca de un sitio de restricción que inactiva la enzima, los clones recombinantes dan una reacción negativa que se traduce en una colonia blanca en contraposición a las no recombinantes que expresan un color azul.
Construcción de bibliotecas de DNA recombinante
El primer paso hacia la identificación de un gene en particular es digerir el genoma que lo contiene en fragmentos manejables de acuerdo con el vector escogido. Lo deseable es que el gene se encuentre localizado en uno de estos fragmentos. La capacidad de clonación de un vector la cual se define como el tamaño máximo que se puede ligar en un vector sin alterar su capacidad replicativa varía dependiendo del sistema de clonación utilizado. El tamaño manejable oscila, de acuerdo al sistema, pero para los plásmidos es de máximo diez mil pares de bases.
Debido al tamaño relativamente grande de los genomas de muchos organismos, el DNA aclonar se fragmenta en pedazos de tamaño aceptable; esta colección de fragmentos se liga a vectores apropiados con el objetivo de construir una biblioteca de clones recombinantes o geneteca. Dentro de la colección de vectores quiméricos habrá uno que tendrá el fragmento que contiene el gene que se quiere aislar. El problema se centra en la metodología a utilizar en la selección del clon recombinante correspondiente. Para tal fin se han desarrollado varios enfoques experimentales, dentro de los cuales el más comúnmente utilizado es la hibridización de colonias de bacterias recombinantes o de placas de lisis de bacteriófagos recombinantes que han crecido sobre superficies sólidas de agar.
El protocolo de clonación molecular más sencillo es emplear una endonucleasa de restricción que produzca cortes que generan extremos cohesivos tanto en el vector como en el DNA foráneo. El ejemplo clásico es la enzima EcoRI tal como ya ha sido explicado en las seccionesanteriores. Posteriormente las secuencias complementarias de los dos extremos de cada DNA se alinean (puesto que estos extremos son homólogos); después del alineamiento, en las moléculas recombinantes se sellan los huecos dentro del vector quimérico con DNA ligasa de T4 (figura 6). Una ventaja de este procedimiento es que no se pierde el sitio de EcoRI, sirviendo como referencial para el estudio y estimación de tamaño del fragmento clonado.