Édgar Vergara1, NelsonAlvis2, Amileth Suárez3
Palabras clave: neoplasias del colon; detección precoz del cáncer; estadificación de neoplasias; biomarcadores de tumor; marcadores genéticos.
Resumen
En la última década han surgido los tratamientos guiados por el perfil molecular del tumor, con beneficio clínico para los pacientes con cáncer colorrectal avanzado o metastásico. Esta estratificación molecular permite agrupar o individualizar a los pacientes para un óptimo tratamiento de su enfermedad.
Basada en información relevante y actualizada, se presenta una revisión de genes y biomoléculas de las vías de señalización intracelular del receptor del factor de crecimiento epidérmico, involucradas en la carcinogénesis del cáncer colorrectal. Además, se pretende identificar evidencia que soporte el beneficio de utilizar biomarcadores en pacientes con cáncer colorrectal, como factores pronósticos o predictivos para tratamientos biológicos.
Se concluye que existe evidencia científica y, además, aceptación por parte de asociaciones internacionales de oncología clínica, para utilizar la evaluación del estado de los genes KRAS y BRAF en la práctica clínica, por su valor predictivo en el tratamiento del cáncer colorrectal avanzado; mientras que, para los genes NRAS, PIK3CA, PETEN y HER2, la aceptación por consenso de expertos de Europa y Estados Unidos aún no es unánime, para recomendar la evaluación rutinaria de estos biomarcadores predictivos en el cáncer colorrectal avanzado.
Introducción
El cáncer es una enfermedad que implica múltiples etapas de mutaciones genéticas y epigenéticas que siguen una evolución darwiniana 1,2. Por esta razón, el tumor presenta una heterogeneidad celular debido a una mezcla de múltiples poblaciones celulares genotípica y fenotípicamente distintas. Este alto grado de heterogeneidad se ha encontrado tanto en tumores del mismo tipo (intertumoral), entre los pacientes con el mismo diagnóstico clínico, o en un mismo tumor (intratumoral), entre las células tumorales derivadas del mismo paciente 3.
El cáncer colorrectal es uno de los cánceres más común e intensamente estudiados en el mundo, con 1,2 millones de casos nuevos y una mortalidad de 6,0 millones al año 4. A pesar de presentar un patrón molecular heterogéneo, más del 80 % de los casos de cáncer colorrectal esporádico se originan de una lesión precursora. La secuencia de adenoma a carcinoma refleja esta transición de activación secuencial de los oncogenes y la inactivación concomitante de los genes supresores de tumores, APC (Adenomatous Polyposis Coli), KRAS (Kirsten Rat Sarcoma viral oncogene homolog), TP53 (Tumor Protein p53), entre otros 5,6 (tabla 1).
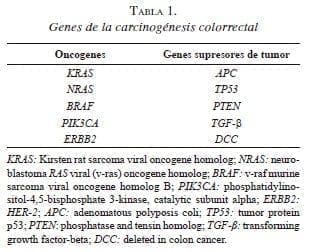
Estos cambios genéticos son cruciales para la progresión tumoral, de modo que la mutación del gen APC es la más temprana y parece ser necesaria para la formación del adenoma de colon; la mutación de KRAS es un evento intermedio y tiene un efecto sinérgico con APC, lo cual se ha observado en, aproximadamente, el 50 % de los adenomas y carcinomas.
Por su parte, la mutación del gen TP53 se ha observado en 75 % de los cánceres colorrectales y participa en la progresión tardía del tumor 7-10. Los eventos anteriores están incluidos en el modelo de Vogelstein, que se ha refinado recientemente por el denominado modelo Big Bang, el cual sostiene que, después de una mutación oncogénica inicial, prevalece una heterogeneidad intratumoral, la cual es favorecida por el desarrollo de subpoblaciones celulares que expresan otros perfiles de mutación oncogénica 11. Las mutaciones en el gen BRAF (v-Raf Murine Sarcoma Viral Oncogene Homolog B) son ejemplos de tales eventos oncogénicos y se encuentran en el 10 %, aproximadamente, de los pacientes con cáncer colorrectal 12,13; y son mutuamente excluyentes con las mutaciones en KRAS/NRAS (v-ras neuroblastoma RAS viral oncogene homolog) 14.
Otros genes alterados en el cáncer colorrectal incluyen PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha), PTEN (phosphatase and tensin homolog) y HER2 (erb-b2 receptor tyrosine kinase 2). La mutación de PIK3CA que surge tarde en la tumorogénesis, justo antes o coincidiendo con la invasión tumoral, se asocia significativamente con la mutación de KRAS y está presente en 10 a 20 % de los casos de cáncer colorrectal 15-17. El gen PTEN es un gen supresor de tumor, su pérdida de función deja en libertad la vía de las proteínas PI3K/AKT (phosphoinositide 3-kinase / protein kinase B), favoreciendo la supervivencia y la metástasis de células tumorales del cáncer colorrectal 18-20. A diferencia del estado del gen KRAS, la expresión de PTEN solo presenta concordancia en 60 % en el tumor primario y sus metástasis, debido a que la pérdida de expresión del gen es más frecuente en las metástasis a distancia 21. El oncogén HER2 presenta mutaciones somáticas o amplificación en 7 % de los pacientes con cáncer colorrectal; esto origina una transformación oncogénica de las células epiteliales del colon, generando resistencia a los tratamientos biológicos 22-24.
El receptor del factor de crecimiento epidérmico (Epidermal Growth Factor Receptor, EGFR), es un receptor tirosina cinasa unido a membrana y se reconoce como un actor importante en la oncogénesis colorrectal, ya que su estimulación por ligandos inicia la cascada de transducción de señales dentro de la célula que culmina activando factores de transcripción que, a su vez, activan genes específicos de la carcinogénesis 25. Presenta dos vías principales de señalización intracelular que controlan la proliferación, la supervivencia y la motilidad celulares 26 (figura 1).
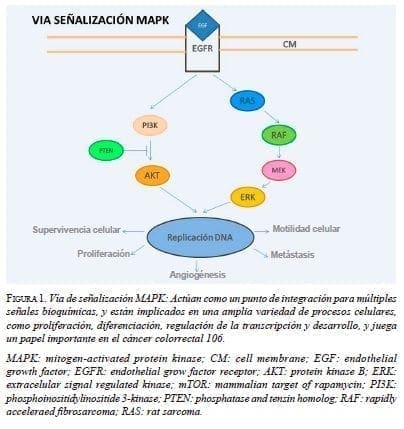
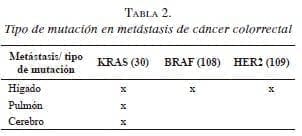
La proteína KRAS participa como una molécula efectora responsable de la transducción al núcleo de las señales de ligandos unidos al EGFR dentro de la vía de señalización MAPK (Mitogen-Activated Protein Kinase) y puede estar alterada en el cáncer colorrectal avanzado o metastásico (metastasic Colorectal Cancer, mCRC) 27. La aparición de metástasis está favorecida por rasgos genéticos de la célula tumoral circulante y por un microambiente tisular favorable en el órgano blanco 28. La mutación del gen KRAS se ha asociado con un mayor riesgo de recidiva pulmonar y supervivencia baja, después de la resección curativa de metástasis hepáticas 29,30. De igual manera, el patrón de alteración de otros genes específicos en el tumor primario del cáncer colorrectal determina el sitio anatómico de aparición de las metástasis 31 (tabla 2).
El EGFR, un receptor tirosina cinasa (Receptor Tyrosine Kinases, RTK), se ha convertido en un objetivo clave de las estrategias terapéuticas diseñadas para el tratamiento del cáncer colorrectal metastásico, en particular, con anticuerpos monoclonales (Monoclonal Antibodies, mAbs) contra su dominio extracelular. El incremento de número de copias de EGFR o mutaciones de los genes responsables de las proteínas de la vía MAPK/ERK (Extracellular signal-Regulated Kinases), en especial la proteína KRAS, son determinantes de la reacción o resistencia a los mAbs-EGFR 32.
Por otro lado, se acepta que en el inicio de la carcinogénesis del cáncer colorrectal se presentan fenómenos de inestabilidad genómica o epigenética, que incluyen: inestabilidad cromosómica (Cromosomal Instability, CIN); inestabilidad de microsatélites (Microsatellite Instability, MSI) fenotipo de metilación de islas de CpG (Cytosine-Phosphate-Guanine), (CpG Island Methylator Phenotype, CIMP); e hipometilación global del ADN 33.
La inestabilidad cromosómica consiste en variaciones que alteran el número normal de cromosomas o que producen cambios estructurales de los mismos, deleciones o translocaciones, produciendo modificaciones en el complemento cromosómico celular 34. Esto lleva a aneuploidía, que es muy frecuente en células cancerosas. La inestabilidad cromosómica puede generarse por mutaciones individuales en un gen concreto, por pérdida o ganancia de cromosomas, o por reorganizaciones a gran escala de los cromosomas 35.
La secuencia de eventos se resume así: la aneuploidía se desarrolla progresivamente a partir de una célula diploide, luego hay pérdida de genes que controlan la proliferación celular, se genera un caos cromosómico continuo que resulta en inestabilidad cromosómica, la cual favorece el desarrollo de tumores 36.
En cuanto a la inestabilidad de microsatélites (MSI), estos son secuencias repetitivas de nucleótidos que existen en el ADN en condiciones normales, pero son muy propensas a presentar mutaciones, que habitualmente son reparadas por los genes reparadores de errores 37. Cuando estos genes se hallan inactivados, se produce el fenómeno de la inestabilidad de microsatélites, y estos aumentan o disminuyen su longitud en la línea germinal 38. Los microsatélites son particularmente propensos a errores en la replicación, debido a que su estructura repetitiva propicia que la ADN polimerasa “se equivoque” al copiar la hebra molde del ADN 39.
El fenotipo de metilación de islas de CpG (CIMP) genera un proceso reversible de los mecanismos epigenéticos, en el cual se introduce o elimina un grupo metilo en las citosinas presentes en dinucleótidos CpG en el genoma 40. Estos dinucleótidos se concentran en zonas llamadas islas de CpG y, frecuentemente, se localizan en los promotores génicos; aproximadamente, el 60 % de los genes humanos contienen islas de CpG en la zona 5’ de su promotor y la metilación de cada grupo celular se fija desde la etapa embrionaria 41.
Se ha reportado que, en el cáncer, los patrones de metilación del ADN sufren modificaciones 42. Si se aumentan, se produce hipermetilación que se traduce en represión de la transcripción génica; por el contrario, si se disminuyen, se produce hipometilación que activa la transcripción. La cantidad de hipometilación del genoma se correlaciona con alteraciones cromosómicas que afectan la estabilidad de cromosomas, los mecanismos de impronta genética y la activación de genes silenciados 43. En general, el ADN tumoral está hipometilado; es por eso que su hipermetilación puede afectar genes específicos que participan en la carcinogénesis. La hipermetilación de los promotores génicos pertenecientes a genes supresores de tumor interrumpen la transcripción, facilitando el desarrollo tumoral 44,45.
En oncología se analizan muchos factores que influyen sobre el tumor, por ejemplo, los denominados biomarcadores tumorales. Estos factores se han clasificado en factores pronósticos y predictivos. El factor pronóstico se define como cualquier parámetro evaluado al momento del diagnóstico (o cirugía), que está asociado con el resultado al final del tratamiento (supervivencia libre de enfermedad, supervivencia libre de progresión, supervivencia global). El factor predictivo es cualquier parámetro que evalúa la mejoría o la falta de mejoría con un tratamiento específico.
En la última década han surgido los tratamientos guiados por el perfil molecular del tumor, con beneficio clínico para pacientes con cáncer colorrectal avanzado 46. Es por eso que, en busca de una evaluación biomolecular más específica y profunda, en los ensayos clínicos se ha incluido la valoración de diferentes genes implicados en su oncogénesis, como KRAS, NRAS, BRAF, PIK3CA, PETEN y HER2; al igual que el estado de reparación del mal apareamiento del ADN 47-51. Esta estratificación molecular permite agrupar o individualizar a los pacientes para un óptimo tratamiento de su enfermedad.
En esta revisión se muestra la participación de diferentes genes que son actores fundamentales en la oncogénesis del cáncer colorrectal, algunos aspectos clínico-patológicos, y la utilidad clínica del estado de los genes que influyen como factores pronósticos, predictivos o ambos de la enfermedad.
Bases moleculares de la oncogénesis en cáncer colorrectal
Los mecanismos moleculares en la secuencia de un adenoma de colon a un carcinoma han sido extensamente estudiados. Estos incluyen múltiples mutaciones en genes supresores de tumor, como APC, DCC (Deleted in Colorectal Cancer protein), TP53 y oncogenes como KRAS que se traduce en inestabilidad genómica 6,52,53. En la progresión tumoral de un adenoma convencional, el KRAS juega un papel fundamental al intervenir tempranamente en el desarrollo del adenoma; además, tiene un efecto sinérgico con la pérdida de función del gen APC, provocando un aumento en el número, el tamaño de la capacidad invasora de los adenomas, y promoviendo la expansión de células madre putativas dentro del epitelio tumoral 54,55. Asimismo, la proteína BRAF, que es un efector de la proteína KRAS y que pertenece a la vía de señalización MAPK/ERK, produce alteraciones de la división y la diferenciación celulares del adenoma de colon 56.
Por otro lado, la activación del gen PIK3CA es responsable de coordinar funciones de proliferación y migración celular mediante las proteínas de la vía de señalización PI3K-PTEN-AKT 57. Cuando hay mutaciones en genes de la familia RAS, ya sea KRAS o NRAS, se producen cambios en un nucleótido que implican producción de un aminoácido diferente y una proteína transformada, la cual pierde su función de GTPasa (enzyme guanosine triphosphate) 58,59. En este caso, se dice que la vía de señalización MAPK estaría constitutivamente activada, porque no depende de la estimulación de ligandos del EGFR, y el resultado es la progresión del proceso de oncogénesis 60.
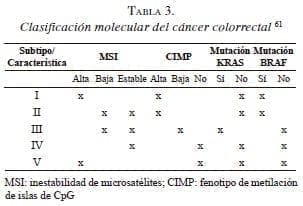
El conocimiento de las mutaciones en los genes KRAS y BRAF, además de la inestabilidad de microsatélites del ADN y del CIMP permitió clasificar el cáncer colorrectal en cinco subtipos moleculares 61 (tabla 3). Posteriormente, se modificó esta clasificación y se incluyeron solo tres de sus vías originales, los grupos I, II y IV, y se utilizó para casos de cáncer colorrectal esporádico 62. En 2012, apareció otra propuesta de clasificación molecular que incluye las vías, Wnt (Wingless-related integration site) y TGF-β (Transforming Growth Factor beta), RTK-RAS y PIK3CA, y, por último, la vía de p53 5. En otra clasificación del 2013, se empleó el análisis multivariado y se pudieron incluir más vías implicadas en la carcinogénesis, entre ellas, NRAS 63.
Por otro lado, en el 2015, en un estudio basado en patrones de metilación de promotores del ADN, se establecieron subgrupos de pacientes que comparten características clínico-patológicas similares y que permiten una nueva caracterización molecular de los tumores en cáncer colorrectal 64. En el 2016, para poder unificar tantas clasificaciones, se elaboró una clasificación de consenso de los diferentes subtipos moleculares del cáncer colorrectal 65.
Combinaciones de mutaciones genéticas
Las mutaciones del gen PIK3CA se encuentran con mayor frecuencia en tumores con el gen KRAS mutado; por el contrario, las mutaciones de HER2 se asocian con el gen KRAS nativo y, en los análisis combinados que incluyen los genes KRAS, BRAF y HER2, las mutaciones de BRAF o las amplificaciones del HER2 se asocian con un mal pronóstico en el grupo del gen KRAS nativo 66,67.
Más del 95 % de las mutaciones del gen KRAS se producen en los codones 12 y 13 del exón 2; otras mutaciones, menos del 5 %, están en los codones 61 y 146, pero no tienen significancia clínica o pronóstica diferente a la presentada por el KRAS mutado en los codones 12 o 13 68,69. También, se han descrito dos mutaciones en el gen KRAS, raras, que afectan el sitio de unión del fosfato en los codones 10 a 16 y producen una alteración funcional. Además, existen variantes de inserción que inhiben la hidrólisis de GTP y favorecen la acumulación celular de RAS activa (RAS-GTP) que, de una manera constitutiva, mantienen las proteínas de la vía MAPK/ BRAF activadas; esto contribuye a la resistencia del cáncer colorrectal avanzado contra el tratamiento con mAbs-EGFR 70.
En adenomas de tipo serrado del colon, los genes KRAS y BRAF actúan como iniciadores de la carcinogénesis y se ha sugerido que la gran actividad de las proteínas de la vía Wnt/β-catenina favorecen la progresión maligna del adenoma serrado en presencia de mutaciones de BRAF, por mecanismos aún desconocidos 71. Por otro lado, en presencia de BRAF en estado nativo y mutaciones de KRAS en su codón 12, hay mayor mortalidad que en casos de mutación en el codón 13 72. La mayoría de las mutaciones del gen BRAF están presentes en los tumores con KRAS nativo 73.
Gen KRAS y clasificación TNM
La clasificación clínico-patológica basada en el sistema TNM ha sido de gran utilidad para pronosticar los resultados clínicos en el cáncer colorrectal 74. Esto ha servido, también, para elegir los tratamientos adyuvantes en el control de la enfermedad 75. Sin embargo, en los últimos años, herramientas moleculares como el estado del gen KRAS tumoral, han permitido predecir e identificar pacientes que se van a beneficiar de la utilización del tratamiento biológico 76,77. Los pacientes con cáncer colorrectal en estadio clínico I y II, en seguimientos de más de seis años, presentan diferentes curvas de supervivencia; al relacionarlas con el estado del KRAS, esta supervivencia ha sido menor en casos de mutación del gen KRAS en el codón 13 78.
Las mutaciones en los genes KRAS, BRAF y NRAS no tienen gran valor pronóstico en el cáncer colorrectal en estadio clínico II y III; al contrario, las mutaciones de PIK3CA sí se han asociado con recurrencia del tumor y serían un factor independiente de mal pronóstico al evaluar el resultado en la supervivencia global 79. Sin embargo, las mutaciones en el NRAS pronostican una menor supervivencia del paciente y, además, podrían tener una influencia predictiva negativa en el caso de tratamiento con mAbs-EGFR en cáncer colorrectal avanzado 80-82.
Por otro lado, el conocimiento del estado de mutación del gen KRAS y el estado de reparación del mal apareamiento (MisMatch Repair, MMR) del ADN, pueden mejorar la estadificación clínico-patológica molecular en el cáncer colorrectal y, además, permiten pronosticar la progresión de la enfermedad metastásica 83.
1 Médico, cirujano oncólogo; director, Grupo de Investigaciones Clínicas en Medicina, Universidad de Sucre, Sincelejo, Colombia
2 Médico, MPH, PhD; director, Grupo de Economía de la Salud, Universidad de Cartagena, Cartagena, Colombia
3 Química farmaceuta, MsC, PhD; directora, Grupo Prometeus, Universidad de Cartagena, Cartagena, Colombia
Fecha de recibido: 3 de octubre de 2016
Fecha de aprobación: 1 de diciembre de 2016
Citar como: Vergara E, Alvis N, Suárez A. Perfil molecular en cáncer colorrectal. Rev Colomb Cir. 2017;32:45-55.Utilidad de la valoración del estado de los genes en la práctica clínica
Los pacientes con cáncer colorrectal en estadio clínico IV, que no han mejorado con la quimioterapia y con el gen KRAS tumoral nativo, se benefician de tratamientos con mAbs-EGFR. Los beneficios se traducen en mejoría de la supervivencia global, supervivencia libre de progresión y conservación de la calidad de vida 84,85. En presencia de KRAS mutado, no se presentan estos beneficios, independientemente del tipo de codón mutado 86.
La mutación del gen KRAS se comporta como un factor pronóstico negativo para la supervivencia global en el cáncer rectal y para la supervivencia libre de enfermedad en el cáncer de colon en estadio clínico II 87-89; de la misma manera, la mutación en el BRAF tiene un efecto pronóstico negativo en la supervivencia global en el estadio clínico II 90,91. Las mutaciones en los genes KRAS y BRAF se asocian significativamente con la supervivencia libre de enfermedad y la supervivencia global más corta en pacientes con tumores de microsatélites estables, pero no en pacientes con tumores con inestabilidad de microsatélites 92.
El cáncer colorrectal en estadio clínico III con deficiencia de la reparación del mal apareamiento del ADN (dMMR), ubicado en el colon distal o con un estado ganglionar N2, tiene un pronóstico malo; esto, independientemente del resultado adverso por mutaciones de KRAS o BRAF 93,94.
Al igual que para el gen KRAS nativo, se siguen investigando nuevos tratamientos dirigidos al gen BRAF nativo en el cáncer colorrectal metastásico 95. También, la valoración de la expresión de HER2, que se asocia con supervivencia global baja y tiene potencial valor predictivo para el tratamiento biológico en el cáncer colorrectal avanzado, se está incluyendo en las investigaciones de nuevos tratamientos 96,97.
Se está estudiando, también, el gen PIK3CA que se presenta más en el colon proximal, asociado con altos niveles CIMP y mutación en el KRAS y el BRAF nativo, y que funciona como evaluador de pronóstico porque se acompaña de una disminución significativa de la supervivencia global 16. Además de la mutación del gen KRAS, las mutaciones de PIK3CA y PTEN pueden ser predictivas de falta de efectividad del tratamiento anti- EGFR en el cáncer colorrectal metastásico 98,99 (tabla 4). Se reporta que hasta en 24 % de los pacientes con gen KRAS nativo, se presentan mutaciones en los genes PIK3CA o BRAF 100,101.
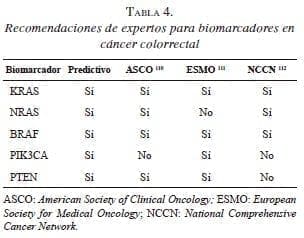
No se ha encontrado asociación significativa entre la mutación de NRAS y la supervivencia de los pacientes con cáncer colorrectal en estadio clínico II y III 79. Sin embargo, la mutación en el NRAS predice resistencia a los tratamientos con mAbs-EGFR en el cáncer colorrectal metastásico 47. De otro lado, el impacto pronóstico del estado de BRAF sigue siendo controvertido 102,103. Sin embargo, las investigaciones continúan para lograr afinar la utilidad clínica de estos biomarcadores 47-49,104, ya que los estudios existentes han demostrado que entre los pacientes con cáncer colorrectal avanzado con el gen KRAS en estado nativo y que albergan mutaciones en los genes de NRAS, BRAF, PIK3CA, o PTEN, se puede demostrar resistencia al tratamiento con mAbs- EGFR 105.
Conclusiones y perspectivas
Se puede concluir que existe evidencia científica y, además, aceptación por parte de las asociaciones internacionales de oncología clínica, para utilizar, en casos clínicos, la evaluación del estado de los genes KRAS y BRAF, por su valor predictivo en el tratamiento del cáncer colorrectal avanzado; mientras que, para NRAS, PIK3CA, PETEN y HER2, la aceptación por consenso de expertos de Europa y Estados Unidos aún no ha sido unánime, para recomendar la evaluación rutinaria de estos biomarcadores predictivos en el cáncer colorrectal avanzado. Se espera el respaldo de más ensayos clínicos para consensuar globalmente este panel de biomarcadores que permitan de forma dinámica controlar la progresión de la enfermedad.
Is it clinically worthwhile to evaluate the state of the KRAS, NRAS, BRAF, PTEN, and HER2 genes in patients with colorectal cancer?
Abstract
Therapies guided by the molecular profile of the tumor have emerged in the last decade, with clinical benefit for patients with advanced or metastatic colorectal cancer. This molecular stratification allows patients to be grouped or individualized for optimal treatment of their disease. Based on relevant and up-to-date information, a mini-review of genes and biomolecules of epidermal growth factor receptor intracellular signaling pathways involved in the carcinogenesis of colorectal cancer is presented. In addition, we intend to identify evidence supporting the benefit of using biomarkers in clinical scenarios of colorectal cancer as prognostic or predictive factors for biological therapies. It is concluded that there is scientific evidence and also acceptance by international associations of clinical oncology to use the evaluation of the state of the KRAS and BRAF genes in clinical scenarios because of its predictive value in the treatment of advanced colorectal cancer, while for the NRAS, PIK3CA, PETEN and HER2 genes the consensus of experts from Europe and the United States of America to recommend the routine evaluation of these predictive biomarkers in advanced colorectal cancer has not yet been unanimous,.
Key words: Colonic neoplasms; early detection of cancer; neoplasm staging; biomarkers, tumor; genetic markers.
Referencias
1. Hanahan D, Weinberg RA. Hallmarks of cancer: The next ge¬neration. Cell. 2011;144:646-74.
2. Choi JD, Lee JS. Interplay between epigenetics and genetics in cancer. Genomics Inform. 2013;11:164-73.
3. Jiang Y, Qiu Y, Minn AJ, Zhang NR. Assessing intratumor hete¬rogeneity and tracking longitudinal and spatial clonal evolutio¬nary history by next-generation sequencing. Proceedings of the National Academy of Sciences of the United States of America – PNAS. 2016;113: E5528-37. doi: 10.1073/pnas.1522203113.
4. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. International J Can. 2015;136:E359-86.
5. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330-7.
6. Fearon ER, Vogelstein B. A genetic model for colorectal tumo¬rigenesis. Cell. 1990;61:759-67.
7. Cho SH, Park SM, Lee HS, Lee HY, Cho KH. Attractor landscape analysis of colorectal tumorigenesis and its reversion. BMC Systems Biology. 2016;10:96.
8. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Ha¬milton SR, Thibodeau SN, et al. APC mutations occur
early during colorectal tumorigenesis. Nature. 1992;359: 235-7.
9. Rodrigues NR, Rowan A, Smith ME, Kerr IB, Bodmer WF, Gannon JV, et al. p53 mutations in colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America -PNAS. 1990;87:7555-9.
10. Zauber P, Marotta S, Sabbath-Solitare M. Copy number of the adenomatous polyposis coli gene is not always neutral in spora¬dic colorectal cancers with loss of heterozygosity for the gene. BMC Cancer. 2016;16:213.
11. Sottoriva A, Kang H, Ma Z, Graham TA. A Big Bang model of human colorectal tumor growth. 2015;47:209-16.
12. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934.
13. Kim TM, Jung SH, An CH, Lee SH, Baek IP, Kim MS, et al. Subclonal genomic architectures of primary and metastatic colorectal cancer based on intratumoral genetic heterogeneity. Clin Cancer Res. 2015;21:4461-72.
14. Fransen K, Klintenas M, Osterstrom A, Dimberg J, Monstein HJ, Soderkvist P. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis. 2004;25:527-33.
15. Nosho K, Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, Zepf D, et al. PIK3CA mutation in colorectal cancer: Relationship with genetic and epigenetic alterations. Neoplasia (New York, NY). 2008;10:534-41.
16. Rosty C, Young JP, Walsh MD, Clendenning M, Sanderson K, Walters RJ, et al. PIK3CA activating mutation in colorectal carcinoma: Associations with molecular features and survival. PLoS ONE. 2013;8:e65479.
17. Ogino S, Lochhead P, Giovannucci E, Meyerhardt JA, Fuchs CS, Chan AT. Discovery of colorectal cancer PIK3CA mutation as potential predictive biomarker: Power and promise of molecular pathological epidemiology. Oncogene. 2014;33:2949-55.
18. Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004;64:3014-21.
19. Kuramochi H, Nakamura A, Nakajima G, Kaneko Y, Araida T, Yamamoto M, et al. PTEN mRNA expression is less pronounced in left- than right-sided colon cancer: A retrospective observa¬tional study. BMC Cancer. 2016;16:366.
20. Molinari F, Frattini M. Functions and regulation of the PTEN gene in colorectal cancer. Front Oncol. 2013;3:326.
21. Molinari F, Martin V, Saletti P, De Dosso S, Spitale A, Campo¬novo A, et al. Differing deregulation of EGFR and downstream proteins in primary colorectal cancer and related metastatic sites may be clinically relevant. Br J Cancer. 2009;100:1087-94.
22. Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832-41.
23. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508-23.
24. Sobani ZA, Sawant A, Jafri M, Correa AK, Sahin IH. Oncogenic fingerprint of epidermal growth factor receptor pathway and emerging epidermal growth factor receptor blockade resistance in colorectal cancer. World J Clin Oncol. 2016;7:340-51.
25. Lemmon MA, Schlessinger J. Cell signaling by receptor-tyrosine kinases. Cell. 2010;141:1117-34.
26. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Mon¬talto G, Cervello M, et al. Mutations and deregulation of Ras/ Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3:954-87.
27. Oikonomou E, Koustas E, Goulielmaki M, Pintzas A. BRAF Vs. RAS oncogenes: Are mutations of the same pathway equal? differential signalling and therapeutic implications. Oncotarget. 2014;5:11752-77.
28. Gupta GP, Massagué J. Cancer metastasis: Building a framework. Cell.127:679-95.
29. Tie J, Lipton L, Desai J, Gibbs P, Jorissen RN, Christie M, et al. KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin Cancer Res. 2011;17:1122-30.
30. Vauthey JN, Zimmitti G, Kopetz SE, Shindoh J, Chen SS, An¬dreou A, et al. RAS mutation status predicts survival and patterns of recurrence in patients undergoing hepatectomy for colorectal liver metastases. Ann Surg. 2013;258:619-27.
31. Nguyen DX, Bos PD, Massague J. Metastasis: From dissemina¬tion to organ-specific colonization. Nat Rev Cancer. 2009;9:274- 84.
32. Fleming M, Ravula S, Tatishchev SF, Wang HL. Colorectal carci¬noma: Pathologic aspects. J Gastrointest Oncol. 2012;3:153-73.
33. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterol. 2008;135:1079- 99.
34. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterol. 2010;138:2059-72.
35. Michor F, Iwasa Y, Vogelstein B, Lengauer C, Nowak MA. Can chromosomal instability initiate tumorigenesis? Semin Cancer Biol. 2005;15:43-9.
36. Komarova NL, Wodarz D. The optimal rate of chromosome loss for the inactivation of tumor suppressor genes in cancer. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7017-21.
37. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558-61.
38. Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M. Genomic instability in repeated sequences is an early somatic
event in colorectal tumorigenesis that persists after transforma¬tion. Nat Genet. 1994;6:273-81.
39. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterol. 2010;138:2073-87.e3.
40. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa J-PJ. CpG island methylator phenotype in colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8681-6.
41. Toyota M, Issa JP. CpG island methylator phenotypes in aging and cancer. Semin Cancer Biol. 1999;9:349-57.
42. Sawada T, Yamamoto E, Yamano H-o, Nojima M, Harada T, Maruyama R, et al. Assessment of epigenetic alterations in early colorectal lesions containing BRAF mutations. Oncotarget. 2016;7:35106-18.
43. Kawasaki T, Ohnishi M, Nosho K, Suemoto Y, Kirkner GJ, Meyerhardt JA, et al. CpG island methylator phenotype-low (CIMP-low) colorectal cancer shows not only few methylated CIMP-high-specific CpG islands, but also low-level methylation at individual loci. Mod Pathol. 2008;21:245-55.
44. Kim MS, Lee J, Sidransky D. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev. 2010;29:181-206.
45. Hinoue T, Weisenberger DJ, Lange CPE, Shen H, Byun H-M, van Den Berg D, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271-82.
46. Bando H, Takebe N. Recent innovations in the USA National Cancer Institute-sponsored investigator initiated Phase I and II anticancer drug development. Jpn J Clin Oncol. 2015;45:1001-6.
47. Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK¬3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014;53:852-64.
48. Sforza V, Martinelli E, Ciardiello F, Gambardella V, Napolitano S, Martini G, et al. Mechanisms of resistance to anti-epidermal growth factor receptor inhibitors in metastatic colorectal cancer. World J Gastroenterol. 2016;22:6345-61.
49. Bokemeyer C, Kohne CH, Ciardiello F, Lenz HJ, Heinemann V, Klinkhardt U, et al. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur J Cancer. 2015;51:1243- 52.
50. Chen J, Guo F, Shi X, Zhang L, Zhang A, Jin H, et al. BRAF V600E mutation and KRAS codon 13 mutations predict poor survival in Chinese colorectal cancer patients. BMC Cancer. 2014;14:1-12.
51. Kawakami H, Zaanan A, Sinicrope FA. Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol. 2015;16:30.
52. Arvelo F, Sojo F, Cotte C. Biology of colorectal cancer. Ecan¬cermedicalscience. 2015;9:520.
53. Fodde R, Smits R, Clevers H. APC, signal transduction and gene¬tic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55-67.
54. Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: Implications for disease behavior and prevention. Int J Mol Sci. 2013;14:16365-85.
55. Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, et al. A two step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623-34.
56. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291-310.
57. Georgescu M-M. PTEN tumor suppressor network in PI3K-Akt pathway control. Genes Cancer. 2010;1:1170-7.
58. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333-8.
59. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: Weaving a tumorigenic web. Nat Rev Can. 2011;11:761-74.
60. Fernández-Medarde A, Santos E. Ras in cancer and develop¬mental diseases. Genes Cancer. 2011;2:344-58.
61. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10:13-27.
62. Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterol. 2010;138:2088-100.
63. Domingo E, Ramamoorthy R, Oukrif D, Rosmarin D, Presz M, Wang H, et al. Use of multivariate analysis to suggest a new molecular classification of colorectal cancer. J Pathol. 2013;229:441-8.
64. Choi W, Lee J, Lee JY, Lee SM, Kim DW, Kim YJ. Classification of colon cancer patients based on the methylation patterns of promoters. Genomics Inform. 2016;14:46-52.
65. Muller MF, Ibrahim AE, Arends MJ. Molecular pathological clas¬sification of colorectal cancer. Virchows Arch. 2016;469:125-34.
66. Nam SK, Yun S, Koh J, Kwak Y, Seo AN, Park KU, et al. BRAF, PIK3CA, and HER2 oncogenic alterations according to KRAS mutation status in advanced colorectal cancers with distant metastasis. PLoS ONE. 2016;11:e0151865.
67. Palacio-Rúa KA, Ahumada-Rodríguez E, Ceballos-García H, Muñetón-Peña CM. Análisis genético en APC, KRAS y TP53 en pacientes con cáncer de estómago y colon. Rev Gastroenterol Mex. 2014;79:79-89.
68. Imamura Y, Lochhead P, Yamauchi M, Kuchiba A, Qian ZR, Liao X, et al. Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mu¬tations in colorectal cancer: Cohort study and literature review. Mol Cancer. 2014;13:1-15.
69. Roa I, Sánchez T, Majlis A, Schalper K. Mutación del gen KRAS en el cáncer de colon y recto. Rev Med Chil. 2013;141:1166-72.
70. Tong JHM, Lung RWM, Sin FMC, Law PPY, Kang W, Chan AWH, et al. Characterization of rare transforming KRAS mutations in sporadic colorectal cancer. Cancer Biol Ther. 2014;15:768-76.
71. Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26:570-9.
72. Imamura Y, Morikawa T, Liao X, Lochhead P, Kuchiba A, Yamauchi M, et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin Cancer Res. 2012;18:4753-63.
73. Gonsalves WI, Mahoney MR, Sargent DJ, Nelson GD, Alberts SR, Sinicrope FA, et al. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J Natl Cancer Inst. 2014;106:dju106.
74. Quirke P, Williams GT, Ectors N, Ensari A, Piard F, Nagtegaal I. The future of the TNM staging system in colorectal cancer: Time for a debate? Lancet Oncol. 2007;8:651-7.
75. Casadaban L, Rauscher G, Aklilu M, Villenes D, Freels S, Maker AV. Adjuvant chemotherapy is associated with improved survival in patients with stage II colon cancer. Cancer. 2016;122:3277-87.
76. Marzouk O, Schofield J. Review of histopathological and molecular prognostic features in colorectal cancer. Cancer. 2011;3:2767-810.
77. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757-65.
78. Dinu D, Dobre M, Panaitescu E, Bîrla R, Iosif C, Hoara P, et al. Prognostic significance of KRAS gene mutations in colorectal cancer – preliminary study. J Med Life. 2014;7:581-7.
79. Shen Y, Han X, Wang J, Wang S, Yang H, Lu S-H, et al. Prog¬nostic impact of mutation profiling in patients with stage II and III colon cancer. Scientific Reports. 2016;6:24310.
80. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023-34.
81. Ciardiello F, Normanno N, Maiello E, Martinelli E, Troiani T, Pisconti S, et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol. 2014;25:1756-61.
82. Chang SC, Lin PC, Lin JK, Lin CH, Yang SH, Liang WY, et al. Mutation spectra of common cancer-associated genes in different phenotypes of colorectal carcinoma without distant metastasis. Ann Surg Oncol. 2016;23:849-55.
83. Li W, Zhi W, Zou S, Qiu T, Ling Y, Shan L, et al. Distinct clinicopathological patterns of mismatch repair status in colorectal cancer stratified by KRAS mutations. PLoS ONE. 2015;10:e0128202.
84. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757-65.
85. Dallol A, Buhmeida A, Al-Ahwal MS, Al-Maghrabi J, Bajouh O, Al-Khayyat S, et al. Clinical significance of frequent somatic mutations detected by high-throughput targeted sequencing in archived colorectal cancer samples. J Transl Med. 2016;14:118.
86. Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, et al. Meta-analysis comparing the efficacy of anti-EGFR monoclonal antibody therapy between KRAS G13D and other KRAS mutant metastatic colorectal cancer tumours. Eur J Cancer. 2016;55:122-30.
87. Deschoolmeester V, Boeckx C, Baay M, Weyler J, Wuyts W, van Marck E, et al. KRAS mutation detection and prognostic potential in sporadic colorectal cancer using high-resolution melting analysis. Br J Cancer. 2010;103:1627-36.
88. Vilorio-Marqués L, Molina AJ, Diez-Tascón C, Álvarez-Cuenllas B, Álvarez-Cañas C, Hernando-Martín M, et al. Características clínicas, anatomopatológicas y moleculares en casos de cáncer colorrectal según localización tumoral y grado de diferenciación. Rev Col Cancerol. 2015;19:193-203.
89. Vargas C, Carranza H, Otero J, Cardona AF. Uso de bevacizumab en pacientes con cáncer de colon metastásico en el Instituto Na¬cional de Cancerología: una serie de casos. Rev Col Cancerol. 2013;17:53-4.
90. Vogelaar FJ, N van Erning F, Reimers MS, van der Linden H, Pruijt H, C van den Brule AJ, et al. The prognostic value of microsatellite instability, KRAS, BRAF and PIK3CA mutations in stage II colon cancer patients. Mol Med. 2015;21:1038-46.
91. Roa I, Game A, Bizama C, Schalper K. Mutación del gen BRAF en pacientes con cánceres de colon y recto con KRAS no mutado. Rev Méd Chil. 2014;142:55-60.
92. Taieb J, Zaanan A, Le Malicot K, Julie C, Blons H, Mineur L, et al. Prognostic effect of BRAF and KRAS mutations in patients with stage III colon cancer treated with leucovorin, fluoroura¬cil, and oxaliplatin with or without cetuximab: A POST HOC ANALYSIs of the PETACC-8 Trial. JAMA Oncol. 2016:1-11.
93. Sinicrope FA, Mahoney MR, Smyrk TC, Thibodeau SN, Warren RS, Bertagnolli MM, et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Cli Oncol. 2013;31:3664-72.
94. Zhang CM, Lv JF, Gong L, Yu LY, Chen XP, Zhou HH, et al. Role of Deficient Mismatch Repair in the Personalized Mana¬gement of Colorectal Cancer. Int J Environ Res Publ Health. 2016;13:892.
95. Korphaisarn K, Kopetz S. BRAF-directed therapy in metastatic colorectal cancer. Cancer J. 2016;22:175-8.
96. Cushman SM, Jiang C, Hatch AJ, Shterev I, Sibley AB, Nie¬dzwiecki D, et al. Gene expression markers of efficacy and resistance to cetuximab treatment in metastatic colorectal can¬cer: results from CALGB 80203 (Alliance). Clin Cancer Res. 2015;21:1078-86.
97. Takahashi N, Iwasa S, Taniguchi H, Sasaki Y, Shoji H, Honma Y, et al. Prognostic role of ERBB2, MET and VEGFA expression in metastatic colorectal cancer patients treated with anti-EGFR antibodies. Br J Cancer. 2016;114:1003-11.
98. Sood A, McClain D, Maitra R, Basu-Mallick A, Seetharam R, Kaubisch A, et al. PTEN gene expression and mutations in the PIK3CA gene as predictors of clinical benefit to anti-epidermal
growth factor receptor antibody therapy in patients with KRAS wild-type metastatic colorectal cancer. Clin Colorectal Cancer. 2012;11:143-50.
99. Lech G, Slotwinski R, Slodkowski M, Krasnodebski IW. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J Gastroenterol. 2016;22:1745-55.
100. Hurtado C, Wielandt AM, Zárate AJ, Kronberg U, Castro M, Yamagiwa K, et al. Análisis molecular del cáncer de colon esporádico. Rev Med Chil. 2015;143:310-9.
101. Fritsche-Guenther R, Witzel F, Kempa S, Brummer T, Sers C, Blüthgen N. Effects of RAF inhibitors on PI3K/AKT signalling depend on mutational status of the RAS/RAF signalling axis. Oncotarget. 2016;7:7960-9.
102. Lo Nigro C, Ricci V, Vivenza D, Granetto C, Fabozzi T, Mira¬glio E, et al. Prognostic and predictive biomarkers in metastatic colorectal cancer anti-EGFR therapy. World J Gastroenterol. 2016;22:6944-54.
103. Palomba G, Doneddu V, Cossu A, Paliogiannis P, Manca A, Casula M, et al. Prognostic impact of KRAS, NRAS, BRAF, and PIK3CA mutations in primary colorectal carcinomas: A population-based study. J Transl Med. 2016;14:292.
104. Chen D, Huang JF, Liu K, Zhang LQ, Yang Z, Chuai ZR, et al. BRAFV600E mutation and its association with clinicopatho¬logical features of colorectal cancer: A systematic review and meta-analysis. PLoS One. 2014;9:e90607.
105. Liu J, Hu J, Cheng L, Ren W, Yang M, Liu B, et al. Biomarkers predicting resistance to epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer with wild-type KRAS. OncoTargets Ther. 2016;9:557-65.
106. Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6:322-7.
107. Armaghany T, Wilson JD, Chu Q, Mills G. Genetic alterations in colorectal cancer. Gastrointest Cancer Res. 2012;5:19-27.
108. Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623-32.
109. Ooi A, Takehana T, Li X, Suzuki S, Kunitomo K, Iino H, et al. Protein overexpression and gene amplification of HER-2 and EGFR in colorectal cancers: an immunohistochemical and fluorescent in situ hybridization study. Mod Pathol. 2004;17:895-904.
110. Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A, et al. Extended RAS gene mutation testing in metas¬tatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy: American Society of Clinical Oncology provisional clinical opinion update 2015. J Clin Oncol. 2016;34:179-85.
111. van Cutsem E, Cervantes A, Nordlinger B, Arnold D, ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl.3):iii1-9.
112. Simen BB, Yin L, Goswami CP, Davis KO, Bajaj R, Gong JZ, et al. Validation of a next-generation–sequencing cancer panel for use in the clinical laboratory. Arch Pathol Lab Med. 2015;139:508-17.
Correspondencia: Édgar Vergara, MD
Correo electrónico: edgar.vergara@unisucre.edu.co
Sincelejo, Sucre, Colombia.