Reporte de un caso
Jorge Mejía Perdigon, MD.*
Luz Nelly Tobar Bonilla, MD**
* Médico Otorrinolaringólogo Hospital Universitario de La Samaritana. Instructor Asociado Colegio Mayor de Nuestra Señora del Rosario.
** Residente ORL Hospital Universitario de La Samaritana. Colegio Mayor de Nuestra Señora del Rosario.
Resumen
El síndrome de Vogt Koyanagi Harada es una entidad sistémica autoinmune caracterizada por uveítis, hipoacusia, meningitis, alopecia, poliosis y vitíligo. Nosotros reportamos el caso clínico de un paciente con sindrome de Vogt Koyanagi Harada tipo II de 35 años con alteración del epitelio pigmentario de la retina, desprendimiento de retina bilateral, atrofia del nervio óptico, acompañado de hipoacusia neurosensorial derecha, quien respondió a la combinación de corticoide con ciclofosfamida vía oral.
Palabras Claves Síndrome de Vogt Koyanagi Harada, síndrome uveomeníngeo, hipoacusia neurosensorial.
Introducción
El síndrome de Vogt Koyanagi Harada (VKH), síndrome uveomeningeo o síndrome de Uveoencefalitis, es una rara entidad que se caracteriza por una reacción inflamatoria que afecta los órganos pigmentados, especialmente la úvea, el pigmento retinal y en grado variable pares craneales generalmente II y VIII (ocasionando hipoacusia neurosensorial uni o bilateral), meninges y encéfalo, piel y anexos (vitíligo, poliosis, alopecia y canicie) (1-3).
En general la enfermedad se inicia con un episodio febril seguido de uveitis bilateral con coroiditis y neuritis óptica, acompañado de hipoacusia neurosensorial y tinnitus, puede desarrollar además vitíligo, poliosis y alopecia, en ocasiones meningitis o encefalitis (4).
Es un desorden de respuesta autoinmune contra células pigmentadas, y aunque su etiología es aún desconocida se ha descrito destrucción de melanocitos causados por células T citotóxicas CD8+ especialmente en piel, aparato visual y aparato cocleovestibular derivados de la cresta neural, con cierta predisposición genética (4); Hammer en 1974 describió anticuerpos circulantes y linfocitos sensibilizados contra la melanina en sangre periférica y LCR; Gocho en 2001 describió autorreactividad de células T contra la tirosinasa (5); igualmente se ha visto coexistencia con HLA-DR4 y DR53, y clara asociación con determinados haplotipos del sistema HLA DR1 y DR4 (6), y con alelos DQA1 y DQB1 (7); se han involucrado mecanismos alérgicos e infecciones virales (citomegalovirus y virus Epstein Barr).
La incidencia y prevalencia no son bien conocidas, sin embargo, son especialmente susceptibles poblaciones de Asia, India, Marruecos, México y Brasil. En Japón la prevalencia se estima en 15 por millón de habitantes con una incidencia de 6,5 casos por millón. El VKH ocurre principalmente en la tercera y cuarta década de la vida, con mayor incidencia en mujeres, siendo raro en menores de 14 años y con un curso más agresivo.
El síndrome de VKH fue descritó por primera vez en el Extremo Oriente alrededor del año 940 A. C. Por un médico árabe quien describió una enfermedad ocular acompañada de poliosis. En 1906 Vogt y en 1929 Koyanagi describieron un síndrome caracterizado por iridociclitis bilateral, uveítis, meningoencefalitis asociada a vitíligo, alopecia e hipoacusia. En 1926 Harada describió la uveítis bilateral asociada a compromiso del sistema nervioso central y signos dermatológicos, así en 1951 la literatura médica llamó a este sindrome como Vogt Koyanagi Harada (6).
El diagnóstico es clínico por ello se han descrito tres tipos de VKH:
– TIPO I: compromiso ocular sin compromiso de oído o piel.
– TIPO II: hallazgos oculares y al menos una manifestación de oídos o piel.
– TIPO III: signos oculares con dos o más manifestaciones de los otros sistemas ya descritos.
Los tipos I y II en su mayoría tienen una duración menor a un año, el tipo III puede estar activa por más de un año.
A su vez puede ser dividido en tres estadíos:
ESTADIO I: prodrómico, consistente en cefalea, febrícula, dolor orbital profundo, epífora y fotofobia, en ocasiones vitiligo.
ESTADIO II: Oftálmico, caracterizado por uveítis bilateral con visión borrosa, fotofobia, disacusia y meningismo. Pueden aparecer los llamados Nódulos de Koeppe (pequeños nódulos en el borde pupilar), precipitados queratínicos corneales, edema de papila con desprendimiento no hemorrágico de retina. Puede durar este estadio semanas o meses. En este estadio aparecen otras manifestaciones como hipoacusia neurosensorial, compromiso de otros pares como II, III, IV, V, VI y VII, meningoencefalitis, alteraciones cerebelosas y espinales tales como paraparesia, enuresis, encopresis, alteraciones en piel como canicie, poliosis, alopecia y vitíligo, trantornos endocrinológicos y vejiga neurogénica.
El compromiso del VIII par da hipoacusia neurosensorial uni o bilateral que puede acompañarse de tinitus. El componente vestibular da vértigo, nistagmus horizontal, alteración del reflejo óculo-vestibular y alteración de los movimientos oculares del seguimiento lento.
Estadio III. Convalecencia, puede durar varias semanas o meses o ser crónico manifestándose uveítis, disacusia, poliosis, vitíligo y alopecia.
No existen test específicos para confirmar el diagnóstico, sin embargo, tenemos ayudas como LCR, plasma, test electrofisiológicos, examen oftalmológico y examen del VIII par con audiometría y pruebas calóricas vestibulares.
Dentro de los diagnósticos diferenciales encontramos: oftalmitis simpática, corioretinitis, cisticercosis, nocardiosis, toxoplasmosis, oftalmitis por herpes zoster, tumor cerebral, esclerosis múltiple, sarcoidosis, enfermedad de Behcet, meningitis, hipoacusia súbita, e hipoacusias neurosensoriales de otras etiologías (1).
El tratamiento recomendado es con corticoides en altas dosis (8), inmunosupresores como ciclofosfamida, azatioprina, clorambucil, y en algunos casos inmunoglobulinas (9) y terapia fotodinámica (10).
El pronóstico es variable y relativamente benigno, en su mayoría con recuperación visual, aunque algunos pacientes persisten con atrofia coroidea, cataratas, atrofia óptica, opacidad corneal y escleritis, cuando existe compromiso del VIII par la recuperación de la hipoacusia neurosensorial es muy baja pero puede detenerse el deterioro con el tratamiento recomendado; dentro de las secuelas neurológicas pueden existir paresia de músculos extraoculares, psicosis y afasia.
Reporte de Caso
Un hombre de 35 años con cuadro de 20 días de evolución de cefalea frontal gravativa, hipoacusia bilateral, tínitus derecho-visión borrosa y disminución de la agudeza visual. Se encuentra visión OD: 20/200 OI: MM 10 cm, oftalmoscopia con papiledema bilateral, hemorragias en llama, y desprendimiento de retina en la mitad inferior bilateral (Figuras 2 y 3). Hipoacusia neurosensorial confirmada con pruebas audiológicas (Figura 1). Potenciales evocados auditivos de tallo cerebral compatibles con hipoacusia neurosensorial; se hace un diagnóstico de Síndrome de Vogt Koyanagi Harada tipo II, se da manejo multidisciplinario con los servicios de oftalmología e infectología iniciándose azetazolamida, prednisona y ciclopentolato ocular.
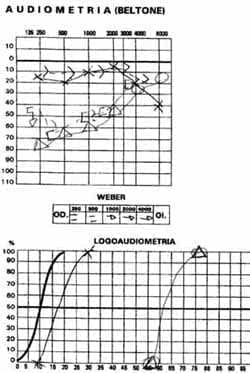
Figura 1. A. Audiometría y logoaudiometría mostrando hipoacusia neurosensorial derecha moderada con pta 55 y oído izquierdo dentro de límites normales, logoaudiometría acorde al pta logrando 100% de discriminación.
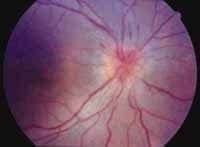
Figura 2A. Angiofluoresceinografía ocular: edema de papila en ojo derecho, atrofia temporal del disco óptico de ojo izquierdo, alteraciones del epr de ambos ojos.
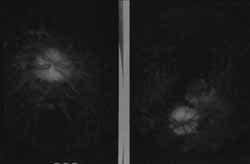
Figura 3. Fotografías de red – free: excavación papilar de 0.0 ojo derecho y de 0.1 en el izquierdo, bordes borrosos en el derecho, no brillo foveolar, máculas hipopigmentadas en ambos ojos.
El paciente mejora su componente oftalmológico con visión OI: 20/80 OD: 20/20, permaneciendo igual compromiso de VIII par, se da salida con corticoide y ciclofosfamida. La evolución ha sido satisfactoria sin deterioro ni compromiso de SNC.
Discusión
El síndrome de VKH es una enfermedad rara cuyo diagnóstico es clínico. El tratamiento de los síntomas es basicamente con corticoides, con mejoría oftalmológica en más del 50% de pacientes según Wendell y Helveston, y mejoría dermatológica según Daisuke; sin embargo, es menos documentada la respuesta a síntomas neurológicos y del VIII par.
En nuestro caso el diagnóstico fue clínico confirmándose con las pruebas audiológicas y con la angioflueoresceinografía. La evolución oftalmológica fue satisfactoria, no así la del VIII par aunque la audición permanece estable.
Siendo una enfermedad poco frecuente por su rareza en nuestro medio, debe tenerse presente como diagnóstico diferencial en pacientes que presentan hipoacusia neurosensorial asociada a trastornos oftálmicos, neurológicos o de piel.
Abstract
Vogt Koyanagi Harada syndrome is an autoimmune systemic disorder characterized by uveitis, meningitis, hearing loss, alopecia, poliosis and vitiligo. We describe a 35 year old patient with Vogt Koyanagy Harada II disease with a retinal pigment epithelium alteration, retinal detachment, optical neuritis, associated with right neurosensorial hearing loss|, who responded to a combination of prednisone and ciclophosphamide oral.
Key words: Vogt Koyanagi Harada syndrome, uveomeningeal syndrome, neurosensorial hearing loss.
Correspondencia: Dr. Luis Jorge Perdigón M. Carrera 16 A # 82-46 consultorio 701 FAX: 5311516: Luz Nelly Tobar B. Calle 98 BIS No. 54-39 int 2 apto. 304. Teléfono: 624 51 80 celular 315-324 42 83. Beeper. 310 55 55 cod 11005.
Bibliografía
1. Araneda L, síndrome de Vogt- Koyanagi- Harada. Cuadernos de Neurología, Chile 1997.
2. Blanco D’ Mendieta JA, Salazar LJA, Síndrome de Vogt Koyanagi Harada en un niño de 4 años. Presentación de un caso clínico. Rev Mex oftalmol 1998; 72: 293-296.
3. Arellanes- García L, Recilas-Gispert C, Síndrome de Vogt Koyanagi Harada. Rev Mex Oftalmol 1998; 72: 59-74.
4. Tsuruta D, Hamada T, Teramae H, Inflammatory vitilgo in Vogt-Koyanagi-Harada disease. Journal of the American Academy of Dermatology. 2001; Volume 44 number 1: 129-31.
5. Gocho K, Identification of autoreactive T cells in Vogt-Koyanagi-Harada disease. Department of Ophtalmology, Akita University School of Medicine, Japan. Invest Ophthalmol Vis Sci 2001 Aug; 42: 2004-9.
6. Siere, Sindrome de Vogt Koyanagi Harada Código CIE 9-MC: 364.24.
7. Chung Hua, Associatin of HLA-DQA1 and DQB1 alleles with Vogt-Koyanagi-Harada syndrome in Han Chinese population. Chinese Academy of Medical Sciences and Peking Union Medical College 1999 may; 35:210-5.
8. Helveston W, Gilmore R, Treatment of Vogt Koyanagi Harada síndrome with intravenous immunoglogulin. Neurology. February 1 1996; Volume 46 Number 2.
9. Latov N, Chaudhry V, Use of Intravenous gamma globulins in a neuroimmunologic diseases. Journal of Allergy and Clinical Immunlogy, October 2001; volume 108 numer 4.
10. Farah ME, Photodynamic therapy with verteporfin for subfoveal choroidal neovascularization in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol 2002 jul; 134: 137-9.