Dr. Alberto Hernández Sáenz
Académico Correspondiente
La Galactosemia, cuyo número de codificación internacional es 271.2, se define como un error del metabolismo de la galactosa, de carácter congénito y es un ejemplo de eugenesia (1), puesto que por medios externos como una dieta, se puede alterar un fenotipo.
Hay creciente interés mundial desde hace unos 20 años por el estudio de los errores congénitos del metabolismo, como la fenilcetonuria y la tirosinemia; han trabajado en ello particularmente en Francia, Brivet; en Inglaterra, ABen; en Suiza, Gitzelmann; Greven en Bélgica; Yonesaba y Ogawa en Japón; Boskowa en Polonia.
En los Estados Unidos hay cuatro centros de investigación sobre estos temas: Harvey Levy y Shi en Bastan; Won Ng en Los Angeles; Weiss en la Academia de Ciencias en Washington y Shulman en el Instituto Nacional de Salud en Bethesda (2) (3).
Los estudios enzimáticos ya se realizan en Colombia, en el Departamento de Bioquímica de la Universidad Nacional (Nancy de Miranda). Existen modernos métodos para detectar fenilcetonuria (PKU) (4) Y galactosemia (5) en recién nacidos por cepas de E. Coli, que se hacen resistentes al bacteriófago C2l en presencia de esta sustancia. La enfermedad fue descrita en Colombia por Hernández (6) en 1983. (Puede interesarle también: Enfermedad de Chagas en Colombia)
HISTORIA DE GALACTOSEMIA
Van Reus (Viena, 1908) (7), describe el primer paciente. Gopper (Viena, 1919) (8), encuentra cuatro niños en una misma familia. (9) Opitz refiere que Fanconi en 1932, expuso en el Congreso de Pediatría de Viena en el año 1932, el caso del niño H.K. de 9 años con cataratas recurrentes, y designa la enfermedad con el nombre de “Galactosa Diabetes”; (10)
Luego de muchos años, en 1980, Gitzelmann (11) lo estudia exhaustivamente cuando ya está ciego y encuentra en el paciente un déficit de galacticol que excluye del diagnóstico de galactosemia.
Lytt menciona que Schwartz (12), en 1955 demuestra el acúmulo de galactosal-fosfato y su toxicidad en eritrocitos de niños galactosémicos, Kalkar en 1956 descubre el error metabólico por la falta de la enzima uridyl transferasa. Beutler (13) y Baluda en 1966, describen un examen sencillo para detectar esta enfermedad en los recién nacidos.
En los años setenta, Levy (14) y Shi practican estudios masivos en cinco millones novecientos mil niños, con el beneficio de la prevención del retardo mental y las graves infecciones que padecen estos niños (15) (16), Merril (17) en 1970, por medio de ingeniería genética, usando un bacteriófago Lambafago Transductor, reemplaza al gene anormal por uno sano.
Fensom (18) en 1979, practica amniocentesis para diagnóstico prenatal. Gitzelmann en el XVI Simposio de Errores Congénitos del Metabolismo, actualiza la secuencia metabólica de la galactosa. Boskowa (19) en el XVI Congreso Mundial de Pediatría, relaciona el nivel de galactosa-I-fosfato y la evolución clínica.
SÍNTOMAS y SIGNOS
Por ser una enfermedad sistémica afecta a todo el organismo. En el recién nacido y en los primeros meses de la vida, la triada de anorexia, vómito y letargia, permite sospechar esta entidad.
Los síntomas más frecuentes de la casuística de McKussick (43 niños) son: anorexia, pérdida de peso, retardo en el crecimiento, hepatomegalia, meteorismo, vómito, emisión de orina oscura, infecciones graves y retardo mental.
GENÉTICA
La galactosemia se hereda en forma autosómica recesiva (20) (21). Los padres son portadores sanos heterocigotes (Fig. 1). De importancia trascendental es demostrar en ambos padres una actividad reducida de la enzima uridyl transferasa. El daño genético parece encontrarse en el cromosoma 9 (Fig. 2), no se sabe si es estructural o regulador, pero sí pleiotrópico (Fig. 2• A).
El cariotipo de los padres es normal.
[enc_su_row][enc_su_column size=”1/2″] Figura No. 1 [/enc_su_column]
Figura No. 1 [/enc_su_column]
[enc_su_column size=”1/2″]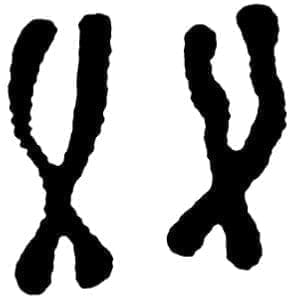 Figura No. 2[/enc_su_column][/enc_su_row]
Figura No. 2[/enc_su_column][/enc_su_row]
 Figura No. 2A
Figura No. 2A
FRECUENCIA
Es de 1 por cada 87.000 en Massachussetts (Levy), aunque Gitzelmann le atribuye 1 por cada 50.000. En Dinamarca, Inglaterra y Austria encuentran frecuencias variables entre 1 por 40.000 y 1 por 80.000.
En América, Europa y Asia el promedio es de 1 por cada 62.000 en una población total de 5’967.089 recién nacidos.
La frecuencia varía según los autores consultados, pues si observamos las cifras de Brennemann en su último libro, publicado en 1980, da una frecuencia de 1 por 18.000.
Los europeos emplean una técnica diferente, al determinar los niveles de Galactosa en sangre, y en esta forma la población afectada se incrementa.
La frecuencia entre nosotros es hasta ahora desconocida. Hernández Sáenz describió los 2 primeros casos en 1983, en trabajo presentado a la Academia Nacional de Medicina.
HISTOLOGÍA HEPÁTICA
Hay escasas necrosis, conformación de pseudo acinos por los hepatocitos, cirrosis incipiente (Fig. 3).
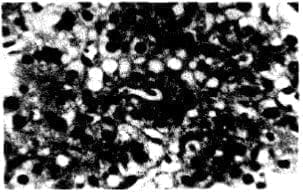 Figura No. 3
Figura No. 3
CONSIDERACIONES BIOQUIMICAS
La galactosemia es ejemplo clásico de un error congénito del metabolismo. El proceso bioquímico que se verifica para que se manifieste dicha entidad se describe a continuación (Bhagavan) (22).
 Figura No. 4
Figura No. 4
Un disacárido como es la lactasa, fuente principal de galactosa en el organismo, se hidroliza por la lactasa en los microvellos del intestino y se transforma en glucosa y galactosa. Posteriormente hay varios pasos metabólicos para entrar en la glucogenolisis como puede observarse en la Figura 5.
 Figura No. 5
Figura No. 5
Se aprecia el bloqueo metabólico por la carencia de uridyl transferasa y la falta de metabolización de la galactosa-l-fosfato que conduce a su acumulación en el organismo.
Algunos pacientes sólo tienen un déficit de galactokinasa y desarrollan únicamente cataratas por producción de D-galacticol como puede observarse en la siguiente reacción:
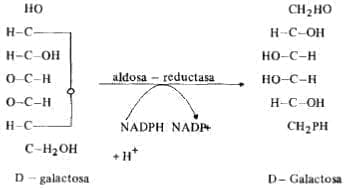 Figura No. 6
Figura No. 6
El diagnóstico de galactosemia se excluye cuando se demuestra el déficit de galactokinasa.
La galactosa se necesita para sintetizar biomoléculas como glucoprotel11as, cerebrósidos, condromucoides y mucoproteínas. Sin embargo el paciente galactosémico con una dieta sin lactosa, es capaz de sin tetizar estas biomoléculas por la reversibilidad de la reacción catalizada por la UDP-galactosa-4-epimerasa.
Esta afirmación ha sido comprobada en linfocitos por Brown y Luis Lelo ir , médico argentino quien demostró en sus célebres estudios sobre la glucogenolisis que las reacciones enzimáticas son de tipo n ucleótido transferasa y generalmente se dirigen a la derecha.
No se ha podido determinar aún si el daño del gene es estructural o regulador, aunque sí se sabe que siempre ocupa el mismo locus.
La molécula de actividad de la enzima uridyl transferasa, tiene un peso molecular de 90.000. Tedesco anota que las mutaciones galactosémicas pueden afectar la síntesis o la degradación de proteínas trasferasa.
Los estudios de patología molecular realizados en Francia por Schapira, describen una familia con la presencia simultánea de gene galactosémico y de la variante Duarte.
PRUEBAS PARA CONFIRMAR EL DIAGNOSTICO
Las múltiples investigaciones que se han realizado desde 1908, han permitido profundizar en el diagnóstico, incluso en el período prenatal, lo que hasta hace pocos años no era factible. Howell (23), afirma que el diagnóstico precoz es posible por amniocentesis y cultivo de células fetales, con líquido obtenido a las doce o catorce semanas de gestación. Los cultivos mencionados se utilizan para análisis citoquímicos.
El niño ictérico con un predominio de bilirrubina indirecta y otros síntomas asociados, puede ser igualmente galactosémico. En el diagnóstico diferencial de proteinurias y aminoacidurias de origen oscuro debe incluirse también esta entidad.
Desde el punto de vista del laboratorio existen dos exámenes fundamentales, uno cuantitativo y otro cualitativo. El más sencillo como tamiz genético es la determinación cualitativa de la deficiencia, en los glóbulos rojos, de la galactosa-l-P. uridyl-trasferasa (24). La muestra se toma por punción capilar en un papel de filtro, como se puede ver en el material utilizado por Levy (Figs. 7 y 8).
 Figura No. 7
Figura No. 7
Figura No. 8
Se impregna totalmente de sangre cada círculo a temperatura ambiente. La muestra por procesar se incuba a 370C, se observa cada hora, durante tres horas, para establecer el grado de fluorescencia, bajo luz ultravioleta de longitud de onda larga.
La intensidad de la fluorescencia depende de la presencia o ausencia de la enzima galactosa-1-fosfato uridyl trasferasa. En portadores heterocigotes, se observa una fluorescencia débil. El examen cuantitativo es una ayuda más en el diagnóstico de la galactosemia congénita, no se usa como procedimiento inicial de diagnóstico, sino como un método de investigación (25).
OBSTACULOS EN EL DIAGNOSTICO: EL PROCESO DIAGNOSTICO DE LA GALACTOSEMIA
Se ha observado que se puede enmascarar porque los síntomas simulan otras entidades tales como enfermedad de Wilson, fructosuria, tirosinemia. Es de vital importancia resaltar este aspecto porque lleva a un diagnóstico errado. Al respecto se refieren varios autores y entre ellos se destacan Donell (26), Pesce (27) y T. Anase (28).
TRATAMIENTO
Como se sabe que el contenido de lactosa en la leche es de 4 a 6% y en el yoghurt de 2 a 3% y que el 40% de la lactosa que éstos alimentos contienen se convierte en galactosa (29), el tratamiento se debe orientar a la supresión de ellos, así como de otros productos que en su metabolismo final conduzcan a la producción de galactosa.
Para prevenir la acumulación de galactosa-1-fosfato y sus metabolitos en los tejidos, es indispensable una dieta estricta carente de lactosa. Se puede sugerir la dieta modelo que aparece a continuación, porque es el resultado de una investigación ejecutada con 41 niños (Figura 9) galactosémicos y durante la cual demostró ser eficiente (30).
 Figura No. 9
Figura No. 9
Por el riesgo de dafio cerebral in útero, se recomienda restringir a las madres de hijos galactosémicos el consumo de leche, porque experimentalmente se ha demostrado la toxicidad de la galactosa transplacentaria en ratas (Spaz y Segall, 1965).
 Figura No. 10
Figura No. 10
Existen opiniones contradictorias sobre la utilización de la leche de soya para el tratamiento de la galactosemia. Schwartz (31) afirma que la leche de soya contiene carbohidratos complejos alfa galactócidos: estaquilosa y rafinosa, los cuales en su metabolismo final se convierten en galactosa generando altos niveles de galactosa-l-fosfato.
Opinión contraria tienen Gitzelmann (32), Brennemann, Bonilla (33), Salazar (34), quienes han demostrado suficientemente que no hay acumulación a niveles peligrosos de galactosa-l-fosfato en eritrocitos.
En el gláfico que aparece a continuación, Brennemann ilustra sus opiniones, contrastando las variaciones que se registran en la concentración de galactosa-l-fosfato, cuando se utilizan diferentes regímenes carentes o no de galactosa, leche de soya y administración de galactosa a dosis variadas.
Finalmente, podemos destacar otras terapéuticas adicionales, que consisten en controlar las infecciones y hemorragias, corregir desequilibrios hidroelectrolíticos, y además buscar el equilibrio emocional del paciente.
PRONOSTICO
Estudios a largo plazo en pacientes galactosémicos han demostrado que la utilización de una dieta libre de lactosa lleva a que el paciente adquiera patrones normales en cuanto al desarrollo físico y psíquico.
Se ha encontrado que las lesiones renales y hepáticas acentuadas, con el tratamiento dietético regresan en cuatro o cinco meses (35).
El tratamiento precoz produce muy buenos resultados con respecto al desarrollo intelectual y de la personalidad. El seguimiento de 45 niños galactosémicos, durante 23 años, le permitió concluir a Fischler (36), que con una dieta carente de lactosa, iniciada precozmente hay desarrollo excelente del intelecto y la personalidad.
Para el seguimiento y control de la dieta se emplean las determinaciones de galactosa-l-fosfato (Gal-l-P), en glóbulos rojos. Boskowa (37) afirma que un nivel de Gal-l-P en eritrocitos por debajo de 40/0 miligramas, demuestra un buen control de la dieta; Tormo (38) obtiene resultados similares.
CONSECUENCIAS
Cuando el diagnóstico o el tratamiento se atrasan, se derivan consecuencias graves como el retardo mental, las cataratas y aún la muerte del paciente.
Nunca el diagnóstico se establecerá por una prueba de tolerancia a la galactosa, pues $e pueden producir acentuadas hipoglicemias y resultados falsos.
PREVENCIÓN
La prevención primaria consiste en el adecuado consejo genético.
La prevención secundaria se hace con dieta precoz sin leche ni sus derivados, y la amniocentesis. (40) Existe una alternativa para la amniocentesis y es la biopsia de las vellosidades coriales.
En los próximos 9 años, siete centros de investigación en U.S.A. se dedicarán a su estudio prospectivo.
Estudios preliminares demuestran su factibilidad, en época precoz como las 6 semanas de gestación, y los resultados son obtenidos en 24 horas.
La amniocentesis tradicional sólo se puede efectuar después de las 16 semanas de vida intrauterina y los resultados se saben luego de cuatro semanas.
Se espera que 4.500 mujeres embarazadas participen en este estudio, con un costo de 2.200.000 dólares.
TRATAMIENTO
Dieta carente de lactosa, con control de los niveles de galactosa-l-fosfato en sangre que no deben ser superiores a 4 miligramos. Si se dan dietas con galactosa, se obtienen niveles altos muy peligrosos: el ideal es el empleo de la leche de soya y una dieta adicional sin lactosa ni galactosa.
CONCLUSIONES
– Se ha demostrado la existencia de galactosemia en Colombia. (39)
– La descripción de la clínica de esta enfermedad debe alertar al médico, sobre todo la tríada anorexia-vómito-Ietargia en el recién nacido.
– Hacer consejería genética adecuada, es la mejor prevención.
– Es la única enfermedad conocida en la cual se contraindica la alimentación materna.
– Las madres galactosémicas portadoras no deben recibir leche durante el embarazo.
– Como la frecuencia de los errores congénitos del metabolismo en Colombia no se conoce, Hernández Sáenz inició un estudio en colaboración con el Doctor Harvey L. Levy, Profesor de la Universidad de Harvard y Jefe del Metabolic Disorders Laboratory de Boston, para investigación a largo plazo de galactosemia, fenilcetonuria, tirosinemia, hipotiroidismo y otros errores metabólicos en niños.
Bibliografía
1. Huekelman et al. Principies of Pediatrics: 349, McGraw Hill Book Company. 1978.
2. Comunicación personal.
3. Comunicación personal
4. Maternal PKU. U.S. Dept. of Health & Human Services. DHS Publications No. (H5A) 81-5299.1982.
5. Levy Paijen, K. H. et al. 1. Lab. Oin. Med. 99: 895: 905, 1982.
6. Hernández S. Alberto. Galactosemia en Colombia. Revista Colombiana de Pediatría y Puericultura. Tomo XXXIV-2 13: 46 Abri11983.
7. Von Reuss, A.; Zuckerausscheidung in Sauglingsalter, Wien Med Wschr 58: 799, 1908. Cit Brennemann’s Practice of Pediatrics Vol. I A Chapter 42: 13. Harper and Row Publishers, 1980.
8. Gopper, F. cited by Bell, et al. Galactose Diabetes (galactosemia). A Clinic Pathologic Study of Two Siblings, J. Pedo 36: 427-39. 1950 Cit. Schaffer Diseases of the Newborn: 815 W. B. Saunders Co. 1960.
9. Opitz. Enciclopedia Pediátrica. Ediciones Morata: 255, 1967.
10. Fanconi/Wallgren; Tratado de Pediatría: 9a. edición. Ediciones Morata: 202, 1972.
11. Gitzelmann R. and Hansen R. G Galactose Metabolism, Hereditary Defects and their Clinical Significance. Proceedings of the Sixteenth Symposium of the Society for the Study of Inborn Errors of Metabolism: 71-72. Edited by Burman D., Holton and Pennock MTP Press Límited. Int. Medical Publishers. Lancaster 1980.
12. Lytt I./Gardner. Enfermedades genéticas y endocrinas de la infancia: 950. Salvat Editores. 1971.
13. Beutler E., and Baluda M.A. A Simple Spot Screening Test for Galactosemia. J. Lab. Clin. Med. 68: 137,1966.
14. Shih V. E., Levy, H. et al, Galactosemia Screening of Newborn in Massachusetts. Ncw Engl J. Med. 284: 753-57, 1971.
15. Levy, Harvey et al. Sepsis Due to Escherichia Coli in Neonates With Galactosemia. New Engl J. Med. 297: 823-25,1977.
16. Shurin, S. B. Escherichia Coli Septicemia in Neonates With Galactosemia. New Engl 1. Med. 25: 1403-4. December 22,1977.
17. Merril, C. R. et al. Bacterial Virus Gene Expression in Human Cells. Nature 233: 398. 1971. Cit. Brennemann’s Practice of Pediatrics Vol. 4 Chapter 19: 18-19. Harper and Row Publishers. 1980.
18. Fensom et al. Prenatal Diagnosis of Galactosemia Heterozigote by Fetal Blood Enzyme Assay. British Med J.: 21-22 January 6.1979.
19. Boskowa et al. Ga1actose-1-Phosphate in Red Blood Cells of Galactosemic Patients in the Course of Treatment Memorias XVI Congreso Internacional de Pediatría: 312. Barcelona. 1980.
20. Beutler, E. et al. A New Genetic abnormality Resulting in Ga1actose+Phosphate Vridy 1 Transferase Deficiency. Lancet 1: 353.1965.
21. Beutler, E. Ga1actose-1-Phosphate Vridyl Transferase. Biochemica1s Methods in Red Cell Genetics. Edited by J. Junis Academic Press. N. Y.: 289-305.1969.
22 Bhagavan, N. V. Bioquímica Editorial Interamericana. 234-37,1979.
23. Howel1, Rodney et al. enfermedades genéticas. Consulta. 40: 12-15. Abril, 1977.
24. Sigma Technical Bul1etin. N-195. Nov. 1978.
25. Sigma Technica1 Bul1etin. N-600 VV. March 1979.
26. Donell, George N. Pitfal1s in the Diagnosis of Ga1actosemia J. of Ped. Vol. 83 N 3: 515-16.1973.
27. Pesce, M. A. et al. Prob1ems in the Diagnosis of Transferase and Galactokinase Deficient Galactosemia. Ann. Clin. Lab. Sci. 10 (1): 26-32, Jan. 1980.
28. Anase, T. et al. Combined forms of Ga1actosemia, Mucopo1ysacharidoses and Pentosurias Diagnosed in Childreno Physiologie. (4): 293-300. Oct.-Dec. 1979.
29. Waard, H. Lactose in Yoghurt. The Lancet: 605. March 15.1980.
30. Brennemann’s Practice of Pediatrics. Vol. 1, Chapter 42: 10. Harper and Row Publishers. 1980.
31. Schwartz, V. Letters to the Editor. Ped. 37: 351-32. 1966.
32. Gitze1mann, R., et al. The Handiling of Soya Alpha Galactosides by a Normal and a Galactosemic Child. Ped. 36: 231, 1965. (Letters to the Editor).
33. Bonilla, N. Luis. Comunicación personal. Abbott Lab. Colombia. 1981.
34. Sa1azar, Rafael. Comunicación personal. Lab. Mead Johnson Colombia, 1981.
35. Applebaum et al. Reversibility of Extensive Liver Damage in Galactosemia. Gastroenterology 69: 496-99. 1975.
36. Fishler et al. Intellectual and Personality Development in Children With Galactosemia. Ped. 5 (3): 412-19.1972.
37. Boskowa et al. Ga1actose -1- Phosphate in Red Blood cells of Galactosemic Patients in the Course of Treatment. Memorias XVI Congreso Internacional de Pediatría: 312. Barcelona, 1980.
38. Tormo Caladin et al. Blood Clearance of Galactose: lts Clinical Interest. Exp. Enferm. Apar. 57 (3): 292- 300. March. 1980.
39. Hernández S., Alberto. Galactosemia en Colombia. Revista Colombiana de Pediatría y Puericultura. Tomo XXXVI. Nos. 2: 13-46. Bogotá, Abril, 1983.
40. News and Comment. American Academy of Pediatrics. 35 (11): 7 November 1984.