Experiencias Clínicas
R. A. PESTANA, MD; GJ. ARIZA, MD; A.M. MARTINEZ, MD, SCC; A.M. TATIS DE M., MD; AJ. VELILLA, MD.
Este trabajo fue presentado en el Foro Quirúrgico Colombiano del XXIII Congreso Nacional de la Sociedad Colombiana de Cirugía, en agosto 12 al 15 de 1997 en la ciudad de Santafé de Bogotá, D.C.
Palabras clave: Tumor carcinoide, Ampolla de Vater, Ictericia, Ampulectomía, Operación de Whipple, Células neuroendocrinas, Criptas de Lieberkühn, Síndrome carcinoide.
Se presentan 2 casos diagnosticados e intervenidos en la ESE- Hospital Universitario de Cartagena, ambos de sexo femenino, de 53 y 37 años de edad.
La primera paciente consultó por dolor abdominal, fiebre ocasional, escalofríos, vómitos e ictericia de aparición reciente; la endoscopia digestiva alta y la biopsia hicieron el diagnóstico de duodenitis ulcerada; la serie gastroduodenal mostró una imagen submucosa de 2 centímetros en la unión de la segunda con la tercera porción del duodeno; en la TAC se encontró la misma lesión circunscrita a la ampolla de Vater observándose dilatación de las vías biliares extra e intrahepáticas.
La segunda paciente consultó por dolor epigástrico, náuseas, con melenas e ictericia durante su hospitalización. La endoscopia digestiva y la biopsia reportaron tumor neuroendocrino. En ambas pacientes los hallazgos fueron, lesión circunscrita a la ampolla de Vater. sin encontrar adenopatías mesentéricas ni metilstasis hepilticas, por lo que a las dos se les practicó ampulectomía. El diagnóstico anatomopatológicojite de tumor carcinoide clilsico insular de la ampolla de Vater. Las pacientes se recuperaron sati.liactoriamente y se controlan por la consulta externa. Se realiza revisión de los aspectos clínicos, diagnósticos y terapéuticos más notables de esta patología.
Introducción
El tumor carcinoide es una neoplasia que se origina en las células neuroendocrinas subepiteliaIes normalmente situadas en las criptas de Lieberkühn: también se han descrito en casi todos los órganos del cuerpo (1-7). Este tumor se puede originar en cualquier órgano derivado del ectodermo primitivo (4): histológicamente es similar a las células endocrinas secretoras de hormonas como las de la hipófisis, tiroides, pulmón, páncreas y tracto gastrointestinal, lo que explica el porqué estos tumores ocasionalmente producen gastrina, calcitonina, ACTH y catecolaminas, que a veces se asocian con otras neoplasias endocrinas (1,4). En su gran mayoría son asintomáticos y pueden permanecer así por años, hasta cuando causan síntomas por obstrucción de una víscera hueca o por aparición de un síndrome carcinoide cuando los mediadores que secretan alcanzan la circulación sistémica (4).
El carcinoide corresponde al 55-86% de todas las neoplasias neuroendocrinas del tracto digestivo, siendo los órganos más frecuentemente afectados el apéndice cecal, el intestino delgado, el recto y el estómago (1,4,8). Su incidencia en la población general es de 1,5 por 100.000 habitantes (4).
La localización del tumor carcinoide en la ampolla de Vater es supremamente rara: desde la descripción en este sitio anatómico por Brentano en 1920, han sido reportados en la literatura mundial tan esporádicamente, que actualmente los casos descritos no superan los 90, siendo la revisión más amplia la de Hatzitheoklitos et al en 1994, quien presenta 2 y revisa 71 casos previamente publicados (9).
En la literatura nacional revisada, tenemos los trabajos de Matuk (3), quien revisa 20; De la Hoz y Brieva (4), quienes presentan 29, y Ariza G y Pestana-Tirado R. (1), quienes presentan 1 e informan 11, sumando un total de 60 casos de tumor carcinoide, de los cuales solamente 1 se localizó en la ampolla de Vater en el Hospital Universitario de Cartagena en 15 años, del total de 11 tumores carcinoides revisados por los autores (1). Este último y otro que se presentó en 1996, completan los dos casos que se reportan en el presente artículo. El objetivo de esta publicación es dar a conocer los 2 primeros casos de tumor carcinoide en la ampolla de Vater reportados en la literatura latinoamericana, diagnosticados y tratados en el Hospital Universitario de Cartagena, y revisar la literatura y hacer énfasis en los aspectos clínicos, diagnósticos y terapéuticos más notables.
Materiales y Métodos
Caso clínico 1
Mujer de 53 años, procedente de Arjona (Bol.), quien consultó en agosto de 1995, por fiebre y vómitos; refirió que su cuadro se inició 4 meses antes del ingreso, con dolor epigástrico acompañado de períodos febriles, asociados a vómitos de contenido alimentario, tratada médicamente como hepatitis viral con mejoría parcial; 8 días antes de su ingreso presentó recidiva del cuadro descrito que se acompañó en esta ocasión de ictericia generalizada y pérdida de peso. Como antecedente de importancia refirió que 8 años antes había recibido tratamiento con radioterapia por carcinoma de cérvix. Al examen físico de ingreso lucía consciente, con ictericia en las mucosas y escleras, febril y deshidratada. TA: 140/100; FC, 90 por minuto; FR, 26 por’ minuto; T, 38°C; cardiopulmonar y abdomen, sin alteraciones. Exámenes paraclínicos: hemoglobina 13,5 g/dL; hematócrito 41%; leucocitos 5.800 /mL; fosfatasa alcalina 1.670 U/mL; bilirrubina total 1,15 mg/dL; bilirrubina directa 0,61 mg/dL; amilasa 81 U/mL; nitrógeno ureico 17 mg/dL; creatinina 0,8 mg/dL.
La ecografía del hígado y vías biliares mostró obstrucción biliar extrahepática que sugirió carcinoma periampular. La serie gastroduodenal informó una lesión submucosa entre la segunda y tercera porción del duodeno que podría corresponder a leiomioma o lesión de la ampolla de Vater de inserción más baja (Figuras 1 y 2). En la endoscopia digestiva se observa en la ampolla de Vater una masa que protruye hacia la luz intestinal, de 2 cm de largo cubierta por mucosa normal en una zona ulcerada en otra, sin sangrado ni salida de bilis; se toma biopsia, reportada como duodenitis aguda y ulcerada.
 |
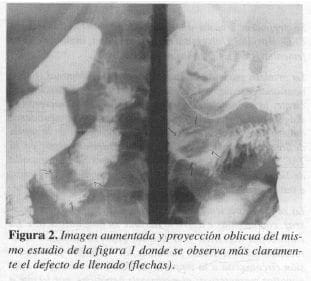 |
La tomografía axial computarizada (TAC) mostró una masa de 2 cm en la segunda porción del duodeno (ampular o periampular) que genera dilatación del árbol biliar y del conducto de Wirsung (Figuras 3 y 4).
 |
 |
Con estos hallazgos la paciente se preparó para cirugía. Se realizó acceso a la cavidad abdominal por incisión de Kocher; se encontró la masa de 2 cm localizada en la ampolla de Vater, ulcerada. Se practicó ampulectomía en razón de la resecabilidad del tumor y de que no se encontraron ganglios comprometidos ni evidencia macroscópica de metástasis hepática. La evolución postoperatoria fue satisfactoria y se ordenó salida del hospital al sexto día postoperatorio.
El reporte de anatomopatología definitivo fue de tumor carcinoide clásico insular de la ampolla de Vater (Figuras 5 y 6).
 |
 |
Caso clínico 2
Mujer de 37 años de edad, procedente de la ciudad de Cartagena de Indias, quien consultó por dolor abdominal y náuseas. Refirió que su cuadro se inició 15 días antes del ingreso, acompañado en sus inicios de melenas y náuseas sin vómitos, que fue tratado como enfermedad acidopéptica. Al momento del ingreso acusó intenso dolor en el hipocondrio derecho y debilidad general. Como antecedente, refirió cuadro similar 12 añós antes; hipertensión arterial diagnosticada 8 años antes y tratada con clorhidrato de propranolol.
Al examen físico se observó una paciente consciente, con palidez generalizada, adolorida e hidratada. TA, 100/90 mmHg; FC, 82 por minuto; FR, 26 por minuto; T: 37.2°C.
Al examen abdominal, acusaba dolor a la palpación superficial y profunda del herniabdomen superior. No se palparon masas. Exámenes paraclínicos, hemoglobina, 5.0 gr/dL, hematócrito, 17%; neutrófilos, 77%; eosinófilos: 3%, linfocitos, 2%; nitrógeno ureico sérico, 6 mg/dL; creatinina, 0,6 mg/dL; bilirubina directa, 0,2 mg/dL; bilirrubina total, 0,6 mg/dL; S-GOT (AST), 22 U/I; S-GPT (ALT), 10 U/I; FA, 171 U/I.
Se transfundieron glóbulos rojos empacados hasta llevar la hemoglobina a 10 g/dL.
La endoscopia digestiva alta mostró la papila ligeramente aumentada de tamaño, eritematosa, (visión frontal). Se realizó examen con visión lateral, observándose papila prominente hipervascularizada, dura al contacto con la pinza; se tomaron biopsias que reportaron neoplasia neuroendocrina que podría corresponder a un carcinoide de la ampolla de Vater. La ecografía y la TAC abdominal no reportaron adenopatías ni metástasis hepáticas. El estudio de serotonina y sus metabolitos urinarios fueron normales (8,65 mg124 horas). Al reinterrogar a la paciente no se halló sintomatología ni signología clínica de síndrome carcinoide. Se realizó acceso quirúrgico subcostal; se encontró la ampolla de Vater de 1,5 cm, sin adenopatías mesentéricas ni metástasis hepáticas, por lo que se realizó ampulectomía más colecistectomía. La paciente se mantuvo estable durante el acto quirúrgico y su posterior evolución fue satisfactoria.
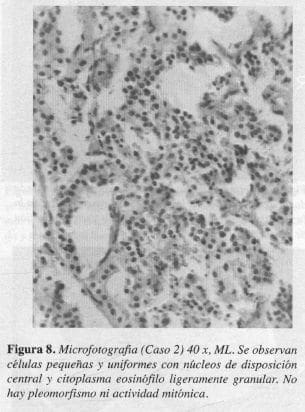
El estudio anatomopatológico definitivo reportó, tumor carcinoide clásico de la ampolla de Vater (Figuras 7 y 8).
 |
 |
Resultados
En ambas pacientes se retiró la sonda nasogástrica al cuarto día postoperatorio y se inició la vía oral con buena tolerancia. Fueron dadas de alta al sexto día postoperatorio. Se realizaron controles por la consulta externa cada 2 meses el primer año y cada 4 meses el segundo año, durante los cuales no han presentado sintomatología y los estudios controles de endoscopia y ecografía abdominales no han reportado alteración patológica.
Discusión
El tumor carcinoide de la ampolla de Vater es una patología extremadamente rara a pesar de ser la neoplasia neuroendocrina primaria más común de la ampolla de Vater (lO). Afecta los dos sexos con una relación hombre mujer de 2: 1; se ha informado su mayor incidencia entre los 23 y 78 años con una media de 51 (9,11).
El tumor carcinoide es la más común de las neoplasias neuroendocrinas del tracto gastrointestinal, representando 55 – 86% de todas ellas, de las cuales, 30 – 60% corresponden al apéndice cecal, 30 – 35% al intestino delgado; 10 – 15% al recto; y existe un 10% reportado en los pulmones (1,4,8); otras localizaciones menos frecuentemente informadas son: estómago, laringe, páncreas, timo, riñón, ovario, próstata, piel, duodeno, e incluso en el apéndice cecal asociado a la endometriosis (1,4,8,12).
No fue nada fácil llegar al conocimiento que se tiene actualmente del tumor carcinoide.
Si se revisa la historia del mismo, nos encontramos con tal número de investigadores que se necesitaría un gran esfuerzo para omitir alguno. Lubarsch en 1888 fue el primero que describió esta lesión en las criptas del íleon distal en la autopsia de 2 pacientes (1, 13). Veintiún años antes Langhan describió un caso al microscopio (l, 14); Ramson en 1860, reportó metástasis hepáticas de un tumor del íleon (l, 15); Kulchiszky, en 1897, identificó células granulares en las criptas de Lieberkühn (15), Giaccio en 1906 describió que estas células tenían afinidad por la plata (l, 5); en 1907 Oberdofer acuñó el término carcinoide (parecido al carcinoma) para referirse a un tumor de procedencia intestinal, pequeño, multicéntrico, de bajo poder invasor e histología característica (16); en 1910 Huebschmann postuló que estos tumores surgían de las células descritas por Kulchiszky en las criptas de Lieberkühn (17).
En 1914 Gosset y Masson fueron los primeros en demostrar que el tumor carcinoide estaba compuesto por células que contenían gránulos intracitoplasmáticos con afinidad por las sales de plata e introdujo el término argentafin (15,18). La naturaleza endocrina de estos tumores permaneció desconocida hasta que en 1948, Rapport et al, aislaron del suero de res, un péptido vasoconstrictor que llamaron serotonina (19), en 1949 Esparmer y Asero concluyeron que esta sustancia se originaba en las células de Kulchiszky y era la misma que ellos llamaban enteramina (1,20); en 1953 Lembeck aisló serotonina de un tumor carcinoide (3,4,15,21); Thorson y Bjork en 1954 describieron el síndrome clínico asociado a los tumores carcinoides (l, 3,4, 15,22) Y en 1955, Page demostró la elevación del ácido 5-0H-indolacético (5HIAA) en la orina de los pacientes .con este síndrome (1,4,15,23,24).
El tumor carcinoide tiene su origen en el sistema neuroendocrino, constituido por todas las células neuronales y endocrinas que comparten un común fenotipo, que consiste en expresar ciertos marcadores proteicos y péptidos; por estose conocen como neoplasias neuroendocrinas (4, 25), originadas estas células de la cresta neura!, tales como las células de Kulchiszky, los melanocitos, células C del tiroides y células cromafínicas de la médula suprarrenal (4); por ello a este grupo de tumores, Bolande los ha llamado neurocrestopatías (26) y Pearse, apudomas (por APUO: Amine Precursor Uptake and Decarhoxylation) (9,27,28), aunque este mismo autor expresa que algunos apudomas pueden surgir de células que no tienen origen en la cresta neural (28). En 1980, la Organización Mundial de la Salud (OMS) clasificó como carcinoides a los tumores que se originaban del sistema neuroendocrino difuso, benignos o con pronóstico más favorable que los carcinomas, caracterizados por un patrón de crecimiento típico, afinidad por sales de plata y reacción inmunohistoquímica positiva con marcadores neuroespecíficos y excreción de diferentes péptidos y aminas biógenas (4). Embriológicamente estos tumores se originan del intestino primitivo y para su clasificación se utiliza la realizada por Willliam y Sandler (Tabla 1) (l, 3, 6, 30).

El intestino medio es el origen del 75% de todos los tumores carcinoides.
Esta clasificación es importante por la variación de las características histoquímicas, propiedades bioquímicas y manifestaciones clínicas del tumor (l, 15).
Las células endocrinas intestinales se clasifican según su reacción histoquímica en (1,6, 31):
A. Argentafines: Dan positividad con la plata (contienen serotonina principalmente).
B. Argirbtllas: Tienen afinidad con la plata pero su reacción es indirecta al adicionar otro reactivo (contienen sustancias peptídicas) .
C. Argirbfobas: No tienen reacción con la plata.
D. Anficrinas: Tienen propiedades mixtas (endocrinas y exocrinas) .
Los tumores que se derivan del intestino anterior son argirófilos mas no angentafines, producen 5-hidroxitriptófano,histamina, ACTH y tienden a causar variantes atípicas del síndrome carcinoide. Los derivados del intestino medio son argirófilos y argentafines, producen serotonina, están asociados con el síndrome carcinode en menos del 10%.
Los derivados del intestino posterior tienden a ser negativos para ambas reacciones, nunca o muy rara vez causan el síndrome carcinoide (1, 3, 6,15,31-33).
El tumor carcinoide típico es un nódulo submucoso pequeño y firme, amarillento, que tiende a invadir hacia la serosa y el borde mesentérico, con una reacción desmoplásica que produce acortamiento del intestino y puede ocasionar episodios de obstrucción intestinal o la presencia de masa (l-4, 15,32, 34).
Las características microscópicas se describen como nidos o cordones de células poligonales monótonas con un núcleo central, esférico, citoplasma granular y eosinófilo. En general, al examen microscópico se debe evaluar el tamaño tumoral, extensión, morfología, agrupamiento célular, según lo propuesto por Soga y Tazawa, aunado a las apetencias tintoriales. Los anteriores autores distinguen cinco tipos o clases de tumores carcinoides (6, 7, 35):
Tipo A: Las células forman nidos sólidos.
Tipo B: Las células se distribuyen en estructuras trabeculares o en cordones que habitualmente, se anastomosan.
Tipo C: Las células se disponen en túbulos, acinos o rosetas.
Tipo D: Existe una escasa diferenciación en las estructuras.
Tipo E: Que también se conoce como tipo mixto, en el cual coexisten elementos de los tres primeros grupos.
Otros autores como Rosai, sólo los dividen en tres categorías que son (l, 36):
a) El tipo clásico, formado por nidos sólidos de pequeñas células monomorfas con ocasional formación de acinos o rosetas; las mitosis son extremadamente raras.
b) Caracterizado por la formación de glándulas sin nidos sólidos, pudiendo haber mucina. La falta de mitosis y atipias, la disposición ordenada y el origen en la base de las glándulas con una mucosa normal sugieren el diagnóstico.
c) Se designa como tumor carcinoide mucinoso; la extensión a músculo y serosa es común, pero como característica la mucosa está respetada, excepto en las áreas de una conexión aparente entre los nidos celulares y la base de las criptas.
El tumor está formado por pequeños nidos uniformes de células en anillo de sello, dispuestas en un modelo microglandular que se acompaña de mucus extracelular. Este tipo de tumor tiene un compartimiento más agresivo que los dos anteriores.
En nuestros pacientes, los hallazgos histológicos fueron compatibles con el tipo A, de la clasificación de Soga y Tozawa y con el tipo clásico, en la clasificación de RosaL en los que se encontró un tumor bien circunscrito pero no encapsulado, donde las células pequeñas y uniformes se disponen formando nidos sólidos y ocasionales acinos; en la profundidad del órgano existe el típico artefacto de retracción entre el tumor y el estroma y la mucosa se observa indemne (Figuras 5, 6, 7 Y 8).
Hay que recalcar que los criterios de malignidad como la anaplasia y las mitosis atípicas, no son aplicables a estos tumores. En ellos las características de malignidad están dadas por la invasión a la serosa, el tamaño mayor de 2 cm, las metástasis ganglionares y a órganos distantes (1, 4).
El tamaño tumoral guarda una relación directa con la enfermedad metastásica, la cual se ha logrado documentar de la siguiente manera: en tumores de 1 cm de diámetro, 2% presenta metástasis; tamaños entre 1 y 2 cm, presentan metástasis un 50% yen tamaños superiores a 2 cm, las metástasis son del 80-90% (l, 8, 32). En la revisión realizada por Hatzitheoklitos, de 71 tumores carcinoides, 46% de éstos iguales o mayores de 2 cm, presentaron metástasis linfáticas o hepáticas (9).
La argentafinidad se puede evaluar con la tinción de Masson- Fontana y la argirofilia con la de Grimelius (7). En nuestros casos no fue posible realizar estas tinciones.
El patrón inmunohistoquímico de los tumores carcinoides de la ampolla de Vater, por ser del intestino anterior, incluye, somatostatina, polipéptidos, neurotensina, serotonina, péptido intestinal vasoactivo (PIV) y calcitonina (9, 11). Se ha informado que esta determinación no tiene influencia pronóstica (9, 11); en muy contadas ocasiones se presenta el síndrome carcinoide y sólo cuando hay metástasis hepáticas (9, 11).
Doctores: Ramiro Alberto l’estalla-Tirado, R-IV de Cirugía General, Jefe de Residentes, Coordinador del Comité de Investigación y Publicación: Genaro .IesÍls Ariza Solallo, R-IV Cirugía General, Miembro del Comité de Investigación y Publicación: Antollio María MartÍllez l’izarro, Jefe Sección de Cirugía General: :\ngela ,liaría Tatis de ,lIorellO, Médica Patóloga, Profesora del Departamento de Patología: Antonio .losé l élilla Rarreto. R-II de Cirugía General, Facultad de Medicina, Universidad de Cartagena, E.S.E. Hospital Universitario de Cartagena, en Cartagena de Indias, Colombia.
Signos y síntomas
La clínica del tumor carcinoide de la ampolla de Vater no es específica y requiere un alto índice de sospecha. El signo más frecuente es la ictericia, la cual se ha encontrado en 62% de los casos (9), tal como se observó en nuestras pacientes; el dolor epigástrico o sensación de plenitud se ha reportado en 40% (9). Puede presentarse melena al ulcerarse y sangrar el tumor, lo que se observó en una de nuestras pacientes. La evolución de los síntomas se ha referido en 3 meses; 1 de las pacientes de este estudio tenía 4 meses de evolución y la otra, 15 días. La edad reportada, como se dijo anteriormente, esta entre 23 y 76 años con una media de 51; en nuestros casos la edad fue de 53 y 32 años, respectivamente; la relación hombre:mujer es de 2: I (9); nuestras 2 pacientes fueron mujeres. Se ha encontrado una relación muy importante entre este tumor y la enfermedad de van Recklinghausen o neurofibromatosis tipo L ya que hasta en un 25% de los pacientes que presentan esta neoplasia, se les ha documentado esta enfermedad (4, 9, 37).
En muy contadas ocasiones se puede encontrar el clásico síndrome carcinoide. La frecuencia es del orden de 2.5% (4, 9), manifestándose con diarreas, crisis de rubor facial, broncoespasmo y afección valvular derecha. Este hecho que se encuentra relacionado con la presencia de metástasis hepáticas, ya que en este caso se evita la barrera depuradora del hígado sobre las sustancias activas secretadas por el tumor. Dichas sustancias ingresan directamente a la circulación general, aunque se ha informado sobre pacientes con metástasis hepáticas múltiples que no presentaron el síndrome carcinoide (7), lo cual podría explicarse por varias razones, entre las cuales tenemos: la reserva hepática para degradar las sustancias secretadas por el tumor y el efecto inhibidor de esta secreción por otras hormonas liberadas localmente; otros autores afirman que puede existir un marcador genético que predispone a ciertos pacientes a presentar o no el síndrome carcinoide en presencia de metástasis hepáticas.
Diagnóstico
Como se ha comentado anteriormente, para el diagnóstico debe existir una gran sospecha clínica, lo que a sido reportado en 15<1< ); en nuestros 2 casos sólo en I se realizó el diagnóstico histopatológico prequirúrgico, debido quizás a las características del otro cuyo tumor tuvo crecimiento hacia la serosa, lo que hace muy difícil realizar una buena biopsia endoscópica, ti menos que la neoplasia esté ulcerada, lo que se ha reportado en 6% de los casos, tal como ocurrió en I de nuestras pacientes. Ayudan en el diagnóstico la imaginología, como los estudios baritados, la ecografía, la TAC, la angiografía y, últimamente, la escintigrafía con octreótida y, además, la endoscopia.
En los estudios baritados del tracto digestivo estas lesiones pueden aparecer como nódulos submucosos, en especial en el duodeno, tal como ocurrió en la serie gastroduodenal del caso I que mostró un defecto esférico submucoso radiolúcido de bordes lisos bien definido y de aspecto benigno, en la unión de la segunda con la tercera porción del duodeno (Figuras I y 2).
La TAC nos informa la localización exacta y, sobre todo, nos descarta las metástasis hepáticas, como se encontró en el caso I (Figura 3). Este estudio debe ser mixto, realizar la fase simple inicialmente y después la contrastada, ya que las metástasis son hipervasculares y se tornan isodensas con el parénquima al infundir el contraste (4).
La angiografía es utilizada para evaluar el manejo terapéutico de las metástasis.
La escintigrafía con octreótida, permite detectar la presencia de receptores de somatostatina; es exitosa en la visualización de la extensión tumoral extrahepática y extraabdominal, encontrándose hasta en 80-95% de lesiones primarias (4,37,38). Cuando la lesión es específica de la ampolla de Vater, la endoscopia nos ofrece una visión exacta (visión lateral) y, dependiendo de la experiencia del endoscopista, se puede tomar una buena biopsia endoscópica que incluya mucosa y submucosa.
En los 2 casos presentados, se realizó endoscopia que mostró papila aumentada de tamaño, eritematosa, hipervascularizada, dura al contacto y en I de ellos, la biopsia fue concluyente.
Como se comentó anteriormente, muy pocos de estos tumores producen el clásico síndrome carcinoide; en la revisión de Hatzitheoklitos, de 71 tumores carcinoides de la ampolla de Vater, sólo 2 casos (2.5%) presentaron síndrome carcinoide clínico y ambos presentaban múltiples metástasis hepáticas (9).
El tamaño tumoral reportado en la ampolla de Vater va de 0.5 a 6 cm, con una media de 2.3 cm (9); en los 2 casos que motivaron este trabajo, los tamaños fueron de 2 y 1.5 cm, respectivamente. El 46% de los tumores de 2 cm o más presentan metástasis linfáticas o hepáticas.
Tratamiento
El tratamiento indicado es la resección quirúrgica, y su extensión está determinada por el tamaño de la lesión y la presencia o no de metástasis (4,9). Se indica la resección local (ampulectomía), en lesiones pequeñas, menores de 2 cm (9, 40), tal como se realizó en los pacientes antes comentados, con lesiones de 2 y 1.5 cm a los que se les hizo ampulectomía, mediante la técnica clásica; la pancreatoduodenectomía (Whipple) se reserva para lesiones mayores de 2 cm, con compromiso ganglionar (9). El pronóstico en general es muy bueno. Se ha reportado una supervivencia quinquenal que supera el 90-95%, y las muertes se relacionan con enfermedad metástasica y progresión tumoral (9). El tumor con metastásis linfática regional macroscópica o con extensión local, debe ser tratado con cirugía agresiva (Whipple); si se puede eliminar toda la enfermedad visible, las tasas de supervivencia a largo plazo serán excelentes. Sin embargo, ciertamente se pueden presentar recurrencias tardías (5-10 años), lo que enfatiza la necesidad del seguimiento prolongado.
En enfermedad metastásica hepática o a distancia, el objetivo es la reducción tumoral mediante resección, embolización arterial hepática, quimioterapia y tratamiento médico con ‘interferón y somatostatina o análogos (4).
La resección es lo ideal cuando afecta áreas hepáticas quirúrgicamente accesibles; de no ser técnicamente posible la resección, se debe realizar la embolización con la que se obtienen buenos resultados ya que estas metástasis se nutren especialmente de sangre arterial (41,42). Los efectos tóxicos producidos por el embolismo son frecuentes y pueden ser severos. Incluyen dolor abdominal, fiebre, náuseas y agravamiento transitorio. Sin embargo, muchos obtienen alivio sintomático subsecuente. Deben realizarse arteriografías previas ya que es preciso tener en cuenta las diferentes variedades de la circulación hepática que se han reportado en estudios realizados en nuestra población (43, 44).
Se ha informado que la infusión de f1uoracilo o doxorrubicina en la arteria hepática, combinada con embolización de la arteria hepática con fibras de colágeno, reducen el volumen de las metástasis del hígado hasta en 50% (45).
La embolización está proscrita cuando existen las siguientes condiciones: tumores que exceden el 50% del volumen hepático, vena porta ocluida, hiperbilirrubinemia o enzimas hepáticas elevadas (4).
El tratamiento médico con interferón alfa puede tener la función de controlar los síntomas del síndrome carcinoide o de detener el crecimiento tumoral, ya que induce la apoptosis celular y favorece la formación de tejido fibrótico (4,46). La combinación de interferón alfa con la infusión continua de f1uoracilo ha demostrado actividad antitumoral y/o antihormonal y, a semejanza de otros regímenes de fármacos, puede proporcionar paliación útil (47).
La quimioterapia actualmente no ha demostrado una excelente respuesta, aunque se ha informado actividad de una variedad de agentes solos y en combinaciones (estreptozocin, doxorubicin, estreptocin, ciclofosfamida, 5-f1uoracilo y clorotocin). Las tasas de respuesta rara vez exceden el 30%. La quimioterapia debe emplearse sólo en forma paliativa en pacientes sintomáticos que deben incluirse en pruebas clínicas orientadas hacia el desarrollo de nuevos tratamientos.
El manejo con somatostatina y sus análogos (octreótida) alivia los síntomas del síndrome carcinoide, con reducción significativa de los niveles de 5-hidroxiindolacético (5-HIAA). Existen agentes como la ciproheptadina que suprimen la producción de aminas vasoactivas o bloquean sus efectos periféricos (4,48-50).
Agradecimientos
Los autores agradecen especialmente a la doctora Lía Barrios, médica patóloga, por la realización de las microfotografías de los especímenes histológicos; al doctor David Scott Jervis Jálabe, por su invaluable colaboración en la búsqueda y revisión bibibliográfica; y al doctor José Luis Puello, Radiólogo, Jefe del Departamento de Radiología e Imágenes Diagnósticas de la Facultad de Medicina de la Universidad de Cartagena, por su valiosa ayuda en el estudio de los pacientes.
Abstract
We discuss two cases of carcinoid tumor of the ampulla of vater diagnosed and treated at Hospital Universitario de Cartagena, Colombia. Both patients were women, 53 and 37 years of age.
Our first patient presemed lI’ith abdominal pain, occasional fe ver, shivers, vomiting, a/1(1icterus oj’recent ouset. Upper gastrointestinal endoscopy and biopsy were informed as ulcerating duodenitis. A gastrointestinal barium contrast studv showed a 2 cm suhmucous mass located at the union 01″ the second a/1(1 third portions 01′ the duodenum. The image lI’as lI’el/ I’isuali;:.ed in CT scan, associated with dilatation oj’hoth the e.rtra- and intrahepatic hilian’ ducts.
The second patinlt consulted hecause 01″ epigastric pain, and nausea, with melenas and jaundice developing during the hospital stay. Endoscopy a11(1hiopsy rel’ealed a neuroendocrine tumor.
At surgical exploration a circunscribed les ion 01′ the ampulla 01′ Vclterwasfound in both j)(ltiell1s: there lI’ere no lnesenteric rdenopathies and no hepatic metastases, and therel’ore a local resection 01″ the (flnpul/a lI’as perl’ormed. Histopathology rel’ealed a c!assical islet cell carcinoid tumor 01′ the ampulla. Both jwtients had an unel’ent/itl recovery and are lI’ell onfiJllolI’-up. We rel’iell’ the clinical, diagnostic, and therapeuticfcaturcs (JI’ this entitv.
Referencias
1. Ariza GJ, Pestana-Tirado RA, Tatis AM: Tumor carcinoide y endometriosis en elapéndice cecal, asociación curiosa. TrihMéd 1997; 95 (5): 2X2-91
2. Pestana-Tirado RA, Ariza GJ, Oviedo L/, Moreno LR: Apendicitis aguda: el diagnóstico es clínico. Trih Méd 1997; 96 (5): 2X2-96
3. Matuk A, Acosta M: Tumor carcinoide. Revisión y casuística. Rev Col CIRUGIA 198X: 3 (2): X3-X9
4. De la Hoz J, Brieva J: Tumores carcinoides y síndrome carcinoide. Rev Col CIRUGIA 1996; II (4): 337-54
5. Fenoglio-I’reiser e. Pascal R, Perzin K: Tumors ofthe intestines. Washington OC .. Puhlished hy the Anned Forces Institute nI’ I’athology 1990
6. Méndez O: Células APUD y apudomas del intestino delgado. Una revisión que enfatiza en tumor carcinoide. Rev Col Gastrocnterol 19X5; 1 (2): 46-54
7. Cantero 1, Gómez M, Maldonado A el al: Estudio de 20 casos de tumores carcinoides del aparato digestivo. Rev Esp Enf Digest 1990: n (5): 2XX-94 X.
8. Delcore R. Friesen SR: Gastrointestinal neuroendocrine tumors. J Am ColI Surg 1994: In: IX7-210
9. Hatzitheoklitos E, Buchler MW, Friess H el al: Carcinoid 01′ the ampulla 01′ Vater. Clinical characteristics and morpho!ogic features. Cancel’ 1994: n (6): 15XO-7
10. Emory RE. Emory TS, Goeilner JR el al: Neuroendocrine ampullary tumors: Spectrum 01′ disease inciuding the lirst report 01′ a neuroendocrine carcinoma 01′ non-small ceil type. Surgery 1994: 115 (6): 763-9
11. Noda y, Watanahe H, Iwuafucim, Furuta K, Ishiharan N el al: Carcinoids and endocrine cell micronests 01′ the minor amI mayor duodenal papilac. Their incidcnce and caracteristics. Cancel’ 1992: 70: 1X25-33
12. Godwin DJ: Carcinoid tumors: An analysis 01′ 2X37 cases. Cancel’ 1′)75: 36: 560-9
13. Luharsh O: Uehcr den primaren krchs des ileum. nehst hemerkungenÍuher das gleichzeilige VOl’kommen von krehs und tuhereulose. Virchow Arch (Path Anal) IXXX:3: 2XO-3
14. Langhans T: Uber eine drusenpoly in ileum. Arch Path Anat 1X67: 3X: 559-66
15. Tilson D: Carcinoid siyndrome. Surg Clin North Am 1974: 54 (2): 409-23
16. Oherndorfer S: Karzinoide. Tumores des dunndanns. Frankfurt Z Path 1907: 1: 420-X 17. Sandcrs RJ, Axtell HK: Carcinoids 01′ the gastrointestinal tracl. Surg Gynecol Ohstet 1964: 119: 369-n
17. Masson P: Careinoids (Argentafin cell tumors) ami nerve hyperplasia 01′ the appendicular mucosa. Am .l. Pathol 192X: 4: 1X1-3
18. Rapport MM, Green AA, Page LH: Partial purification 01′ vasoconstrictor in heef serum . .1Hiol Chem 194X: 147: n5-7
19. Espanner V, Asero B: Identification 01′ enteramine, thc specific honnone ofthe enterochromaffin cell system. as 5-hydroxytryptamine. Nature 1952: 160: XOO-3
20. Lcmheck L. 5-hydroxytryptamine in carcinoid tumor. Nature 1953: 910-13
21. Thorson A, Bjorck G, Bjorkman G, Waldestrom: Malignant carcinoid 01′ the small intestine with metastasis to the liver, valvular disease 01′ the right side 01′ the heart, peripheral vasomotor symptoms. Hronchoconstriction an unusual type 01′ cyanosis. Am Heart.l 1954: 47: 795-XOO
22. Paage lH, Coreoran A, Undenfriend S: Simple test for the diagnosis 01′ the metastatic earcinoid (argentafinoma) eorrcspondence. J Am Mass 1955: 159: 397-9
23. Paage lH, Corcoran AC, Undenfriend S el al: Argentafinoma as an endocrine tumor. Lancet 1955: 1: 19X-9
24. Polak JM: Dianostic histopathology 01′ neuroendocrine tumors. Edinburg, Churchill Livingstone 1993
25. Bolande Rp: The neurocrestopathies: A unifyng concept 01′ the disease arising 01′ neural crest maldevelopmenl. Hum Pathol 1974: 5: 409-29
26. Pearse AG: The APUD concept and hormone production. CJin Endocrinol Metab 19XO:9: 211-22
27. Pearse AG, Polak JM. Heath CM: Polypectide honnone production hy eareinoid apudomas and their relevant cytochemistry. VirchowsAreh (eell pathol) 1974: 16: 95-110
28. Pearse AG, Takor TT: Embriology 01′ the dilTuse neuroendoerine system and its relationship to the common peptides. Fed Proc 1979: 3X: 222X-94
29. Williams E, Sandler M: The ciassification 01′ careionoid tumors. Laneet 1963: 1: 23X-9
30. Méndez O: Péptidos de los sistemas nervioso y digestivo. Las células APUD y su expresión tumoral. Un enfoque básico. Act Méd Colomb 19XX: 13 (4): 248-50
31. Greager JA, Eckhauser MC, Pennington LR: Neoplasias del intestinos delgado. En: Zuidema GD Shackelford, Cirugía del Aparato Digestivo. Tomo V, 3a Ed, Buenos Aires, Editorial Panamericana S.A., 1993. p. 523-49
32. Pei\aloza A, Amaya L, Piña R el al: Tumor carcinoide del estómago. Presentación de dos casos. En: Temas eseogidos de gastroenterología, Tomo XXV, 19X9: 109-16
33. Horsley BL. Baker RR: Fibroplastic response to intestinal carcinoids. Am Surg 1970: 36: 676
34. Soga J, Tazawa K: Pathologie analisys 01′ earcinoids. Histology revaluation 01′ 62 cases. Cancel’ 1972: 2X: 990-8
35. Rosai J: Ackermanís surgical pathology, XI” ed. NY: Moshy Year Book.lnc., 1996
36. Stamm H, Hedinger CE, Sademaslani P: Duodenal and ampullary carcinoid tumors. A report 01′ 12 cases with pathological charaeteristics, polypeptide content and relation to the men Y syndrome and Von RecklinghausenÍs disease. Virchows Arch Pathol Anat Histhopathol 19X6: 40X (5): 475- 89
37. Joseph K, Stapp 1, Reinecke Jet al: Receptor scintigraphy with in-pentetreotid for endocrine gastroenteropancreatic tumors. Horm Metab (suppl) 1993: 27.: 2X-31
38. Lamherts SWJ. Cahyvialle JA, Krenning El’: The visualization 01′ gastroenteropancreratic endocrinc tumors. Metaholism 1992: 41: 111-4
39. Akerstrom G: Management 01′ carcinoid tumors 01′ the stomach, duodenum, and pancreas. World.l Surg 1996: 20 (2): 1n-X2
40. Carrasco CH, Charnsangavcj C. Ajani J el al: The carcinoid syndrome: Palliation hy hepatic artery emholization. Am J Roentgeno119X6: 141 (1): 149-54
41. Moertel CG, May GR, Martin JK el al: Sequential hepatic artery occiusion and chemotherapy for metastatic carcinoid tumor and islet cell carcinoma. Proc Am S ClinOncology 19X5: 4: XO
42. Gonzálc/ A, Pestana-Tirado RA. Ariza GJ, Rangel H el al: Variedades anatómicas de la arteria hepática. Rev Col CIRUGIA 1997: 12(1):3X-45
43. Ariza G1, Pestana-Tirado RA, Rangel H el al: Variedades anatómicas del tronco celíaco. Trib Med 1997: 95 (4): IXI-X
44. Diaco DS. Hajarizadeh H, Mueller CR el al: Treatment 01′ metastatic earcinoid tumors using multimo dality therapy 01′ octreotide aeetate, intra-arterial chemoembolization. Am.l Surg 1995: 169 (5): 523- 8
45. Oherg K, Norheim y, Lind E el al: Treatment 01′ malignant carcinoid tumors with human leukocyte interferon: long-term results. Cancel’ Treat Reports 1986: 70 (11): 1297-304
46. Andreyed HJ, Scott-Mackie P, Cunningham D el al: Phase II study of continuous infusion fluorouracil and interferon alfa-2b in the palliation of malignant neuroendocrine tumors. J Clin Oncol 1995; 13 (6): 1486-92
47. Kvols LK, Moertel CG, Oconnell MJ el al: Treatment of lhe malignant carcinoid syndrome: Evaluation of a long-acting somatostain analogue. N Engl J Med 1986< 315 (11): 663-6
48. Kvols LK, Martín JK, Marsh HM el al: Rapid reversal of carcinoíd crisis with a somatostain analogue. N Engl J Med 1995: 313 (9): 1229-30
Correspondencia:
Doctor Ramiro Alberto Pestana- Tirado. Sección de Cirugía General E.S E. Hospital Universitario de Cartagena, Cartagena de Indias, D. E. Y T.
Comentario
Doctor Jaime A. De la Hoz, Profesor Emérito de la Facultad de Medicina de la Universidad Nacional de Colombia. Ex presidente de la Sociedad Colombiana de Cirugía.
Señor Editor:
Cinco tipos mayores de tumores neuroendocrinos pueden distinguirse en el duodeno: Tumores productores de gastrina; tumores productores de somatostatina; paragangliomas gangliocíticos; tumores que producen serotonina / calcitonina / sustancia PP, y carcinomas pobremente diferenciados. En contraste con los tumores neuroendocrinos del estómago, en los cuales el tamaño del tumor y su asociación con otras enfermedades son el factor pronóstico más importante, el comportamiento de los tumores endocrinos del duodeno está, además del tamaño, íntimamente asociado con el aspecto funcional (1,2). Lo más probable es que los dos casos presentados en este artículo por Pestana-Tirado R A, Y colaboradores, pertenezcan a los productores de somatostatina. Las determinaciones de la serotonina y sus metabolitos urinarios, que se efectuaron en el caso clínico No. 2, fueron normales.
Los carcinoides del duodeno constituyen el 2% de todos los tumores neuroendocrinos del tracto gastrointestinal. Los autores de este trabajo refieren no más de 90 casos en la revisión de la literatura, en la ampolla de Vater. Los estudios han demostrado que los gastrinomas son los tumores neuroendocrinos más frecuentes del duodeno. Del 30 al 40% de los gastrinomas primarios, tienen localización duodenal. Estos tumores son a menudo ocultos a la exploración convencional y su detección requiere duodenotomía y meticulosa evaluación de la mucosa por eversión y palpación directa. Además, pueden ser tan pequeños como de I ó 2 mm, y estar asociados a metástasis a los ganglios linfáticos, los cuales pueden ser considerablemente más grandes que el tumor primario (3). La tercera parte está asociada al síndrome de Zollinger – Ellison (SZE) y los restantes permanecen clínicamente silenciosos y sólo son reconocidos en forma casual, por su intensa inmunorreacción a la gastrina en el espécimen quirúrgico. Aunque los gastrinomas asporádicos son característicamente únicos, los pacientes con síndrome endocrino múltiple (MEN – 1), casi invariablemente tienen múltiples gastrinomas. Aproximadamente 25% de estos pacientes presentan MEN – 1 (4). Independientemente de si los carcinoides son pancreáticos o extrapancreáticos, múltiples o simples, el 90% de ellos se encuentran en un área anatómica específica, designada como el “triángulo del gastrinoma”(5). Los ápices de este triángulo están definidos por la unión del canal cístico y el conducto biliar común, la unión de la segunda y tercera porción del duodeno y la unión del cuerpo con el cuello del páncreas.
Los gastrinomas son generalmente bien circunscritos, pero no encapsulados. Histológicamente no pueden ser distinguidos de otros tumores endocrinos gastroenteropancreáticos y muestran un patrón de crecimiento tubular o trabecular o una combinación de éstos, con una diferenciación tubular prominente (6). Las calcificaciones y la hialinización son aspectos relativamente comunes. Como con todos los otros tumores neuroendocrinos gastroenteropancreáticos, no existe un criterio histológico confiable de malignidad. Esta puede ser demostrada solamente por la presencia de metástasis, infiltración de los órganos adyacentes o invasión de vasos sanguíneos grandes.
El tratamiento del SZE comprende por lo menos tres pasos: el control de la hipersecreción del ácido clorhídrico del estómago, la localización de los tumores productores de gastrina, y la consideración del tratamiento quirúrgico.
Los tumores productores de somatostatina en el duodeno, son segundos en frecuencia y constituyen el 20°!cJ de los tumores neuroendocrinos de este órgano. Ocurren casi exclusivamente en la ampolla de Vater, como ocurrió en los 2 casos que estamos comentando. Histológicamente presentan cuerpos de psammoma. En el momento del diagnóstico, la mayoría muestran invasión de la capa muscular y compromiso de los ganglios linfáticos regionales. Aunque reaccionan intensamente a la somatostatina, no son funcionantes (no se asocian al síndrome de somatostatinoma), como sucede con los pancreáticos. Dependiendo del tamaño del tumor y de la edad del paciente, pueden ser extirpados localmente o por pancreatoduo denectomía.
Los paragangliomas gangliocíticos son raros, ocurren casi exclusivamente en la segunda porción del duodeno, a veces asociados a neurofibromatosis. Están compuestos de una mezcla de paraganglioma, ganglioneurona y tejido carcinoide (7). Son tumores generalmente benignos, reconocidos incidentalmente o debido a sangrado, y tienen un excelente pronóstico después de la resección quirúrgica.
Otros carcinoides duodenales son menos frecuentes y pueden mostrar reactividad a otras hormonas como calcitonina, serotonina y sustancia PP. La mayoría se encuentra en la parte proximal del duodeno y son de bajo grado de malignidad, siendo susceptibles de resección local.
Los autores de este trabajo aprovecharon el hallazgo de 2 casos de tumores carcinoides en la ampolla de Vater, para hacer una juiciosa revisión del concepto carcinoide que hoy rige sobre estos tumores. Enriquecen la casuística de esta localización sumamente rara, y establecen una línea de conducta que está de acuerdo con lo observado en el comportamiento de estas lesiones, es decir, que el tamaño de ella es el que, en la mayoría de las circunstancias, condiciona la extensión de la resección. En este caso, neoplasia menor de 2 cm implica resección local. También es importante anotar la elegante presentación de la embriología, estudios histológicos y métodos dignósticos, que nos permiten ponemos a tono con las tendencias modernas en un tema de mucha controversia en cuanto al verdadero significado de esta patología. Desafortunadamente, todavía en nuestro medio carecemos de la tecnología que nos permita efectuar estudios de inmunohistoquímica que faciliten a los investigadores, qué tumores carcinoides contienen varias sustancias hormonales. Debido a esta deficiencia, es imposible decir a cuál de los grupos tumorales arriba consignados, pertenecen los 2 especímenes de esta presentación.
En conclusión, los autores aportan 2 casos de tumores neuroendocrinos de localización sumamente rara, como es la ampolla de Vater, y contribuyen así a enriquecer la literatura con su estupenda revisión.
Referencias
1. Martín ED. Potet F: Pathology 01′ Endocrine Tumors of the GI Trac!. Clin Gastroenterol 1974; 3: 511-4
2. De la Ho/. J, Brieva J: Tumores Careinoides y Síndrome Carcinoide. Rev Col CIRUGIA 1996; 11 (4): 337-54
3. Pipeleers – Marichal M, Donow C, Heitz P Y, Kloppcl G: Pathologic aspeets 01′ gastrinomas in patients with Zollinger – Ellison syndrome with and without multiple endoerine neoplasia type 1. World J Surg 1993; 17: 4RI-5
4. Andersen D K: What’s New in general surgery. Ann Surg 19R9; 210: 6R5-703
5. Stabile B E, Morrow D J, Passaro E Jr: The gastrinoma triangle: operative implication. Am J Surg 19R4; 147: 25-31
6. Stamm B, Haeki W H, Kloppel G, Heitz P V: Gastrin – produeing tumors and the Zollinger Ellison syndrome. In: Endocrine Pathology of the Gut and Panereas. Edited by 1 Daya. Boca Raton, Florida: CRC Press; 1991
7. Kheirsm, Halpern N B: Paraganglioma 01′ the duodenun in assoeiation with eongenital neurofibromatosis: Possible relationship. Cancer 1984; 53: 2491-4.