J. A. NIETO, MD, SCC, FACS.
Palabras claves: Pancreatitis aguda biliar, Migración del cálculo, Alcoholismo, Lípidos Trauma pancreático Pancreatitis Posoperatoria, Isquemia pancreática, Radicales libres, Colocalización, Lisosomas, Hidroiasas, Crinofagia. ‘
Se presenta una revisión sobre la fISiología de la secreción pancreática y la flSiopatología de la pancreatitis aguda, en la que se analizan las diferentes teorías al respecto; se comenta la migración del cálculo coledociano como causa de la pancreatitis de origen litiásico biliar; las teorías propuestas sobre la obstrucción-hipersecreción y las alteraciones del perfil de lípidos y de la secreción exocrina del páncreas en la pancretitis alcohólica. Se presentan conceptos relacionados con la isquemia y la producción de los radicales libres de oxígeno en la génesis de la entidad y la responsabilidad que éstos pueden tener en la progresión de las formas edematosas a las más severas de necrosis y hemorragia.
Finalmente, se plantea la nueva teoría de la colocalización de las hidrolasas celulares dentro del interior de los lisosomas de manera conjunta con los precursores enzimáticos de la secreción del páncreas y la posibilidad que aparece a la luz de estos nuevos conceptos, de que la lesión inicial en la cadena de eventos bioquímicos que caracterizan a la pancreatitis aguda se presenta intracelularmente y no por lesión de la barrera celular, como hasta el momento se acepta.
Introducción
La pancretitis aguda es una entidad que puede variar de lo trivial a lo fatal; muchos aspectos de la entidad son pobremente comprendidos y, a menudo, objeto de controversia. No existe una clasificación de la pancreatitis y sus complicaciones universalmente aceptada; no hay pleno conocimiento con relación a sus factores etiológicos y sobre el mecanismo mediante el .cual se inicia el proceso fisiopatológico que caracteriza la entidad. En general, existe muy poco conocimiento sobre su incidencia y no hay plena coincidencia entre los estudiosos del tema, con respecto a los índices pronósticos que pueden ser empleados para pronosticar la severidad del episodio. Pero en donde existe una mayor controversia es en los aspectos relacionados con su tratamiento, en particular en la necesidad, momento y extensión de la intervención quirúrgica.
Definición
La primera referencia a la enfermedad inflamatoria del páncreas aparece en 1672, cuando Tulpius describe un caso de absceso difuso del páncreas. En 1799, Baillie informó las características microscópicas de la pancreatitis crónica y aunque la primera observación descrita de un caso de pancreatitis con necrosis grasa fue realizada por Balzer en 1879, poca atención se prestó a la pancreatitis como entidad hasta 1889 cuando Fitz publicó su clásico escrito describiendo el . síndrome clinicopatológico. Precisamente cuando la pancreatitis aguda había sido establecida por Fitz, en 1904 Mayo-Robson produjo su serie de Lecturas Hunterianas en donde describe las características de la pancreatitis crónica y sus complicaciones (1).
Teniendo en cuanta que las primeras descripciones de la enfermedad inflamatoria del páncreas se realizaron a partir de observaciones posmórtem, ésta vino a ser definida y clasificada mediante características anatómicas macroscópicas sin tener en cuenta en muchas ocasiones la extensión y severidad del proceso inflamatorio. Podríamos definir entonces a la pancreatitis aguda como un proceso autolítico-químico, caracterizado por la activación dentro del órgano de enzimas pancreáticas que van a producir lisis y necrosis celular, con liberación de gran cantidad de sustancias tóxicas y vasoactivas que pueden ocasionar, a más de las lesiones locales, múltiples fallas en diferentes sistemas orgánicos.
Fisiologia
Desde el punto de vista funcional es importante revisar los factores que intervienen en la secreción pancreática en cuanto al volumen de producción, composición, síntesis enzimática y electrolítica y transporte de las enzimas de su lugar de producción al canalículo secretor.
Claude Bernard, uno de los primeros estudiosos de la química y la fisiología pancreática, contribuyó al estudio del tema al comienzo de la era moderna de la medicina científica, mediante la aplicación del método experimental a problemas de interés médico. En su obra Indroducción al Estudio de la Medicina Experimental, plantea que: “un adecuado conocimiento de las situaciones patológicas o anormales, no puede adquirirse sin el conocimiento previo Otro tipo de inhibidor de la tripsina, el inhibidor Kunitz, se encuentra presente en el páncreas de bovinos; este inhibidor que tiene una amplia especificidad se ha encontrado en otros órganos bovinos diferentes al páncreas, incluyendo parótida y pulmón. Este tipo de inhibidor no se ha logrado aislar en el páncreas humano; la aprotinina (Trasilol) preparada a partir de parótida bovina es un ejemplo de inhibidor Kunitz (2, 7).
Secreción hidroelectrolítica
En el hombre se producen 25 rnLlk/día de fluido pancreático, con una concentración aproximada de bicarbonato de 100 mEq/L y un pH de 9.0 e iso osmótico con el plasma (9). Se ha implicado a las células ductales en la secreción hidroelectrolítica de la glándula y esta alta concentración de bicarbonato y el pH alcalino, hacen a la secreción pancreática única dentro de los fluidos biológicos. El jugo pancreático es isotónico con respecto al líquido extra celular a cualquier velocidad de secreción y el agua entra en forma pasiva a los conductillos siguiendo gradientes osmóticos, en tanto que la secreción de electrolitos es activa.
Los principales aniones pancreáticos son el Cl- y el HC03-. Los cationes Na+ y K+, son secretados a concentraciones fijas y similares a las del líquido extra celular, la concentración de cloruros se ha encontrado en una relación inversa con la del bicarbonato, manteniendo estable la concentración de aniones que en conjunto totalizan aproximadamente 150 mEq/L, e igual a la de cationes, en donde el sodio y el potasio presentan concentraciones muy similares a las plasmáticas (10).
Mecanismo de la secreción de bicarbonato
De acuerdo con Swanson y Solomon (7) en la secreción de bicarbonato, además de las células ductales, también intervienen las células acinares; en el sistema ductal intralobulillar la secreción electrolítica ocurre de manera espontánea. Según los estudios de Case (4), es mediada por la bomba H+/Na+, estimulada por acción de la secretina en el sistema ductal extralobular, iniciándose por la salida de H+ de la célula ductal al fluido intersticial en intercambio por Na+, esto deja un ion OH- que se combina con el CÜ2 proveniente del metabolismo celular, reacción que se encuentra mediada por la anhidrasa carbónica, y es en el sistema ductal principal en donde las células de la pared del ducto tienen la capacidad de modificar el volumen y composición del fluido producido por las células acinares y ductales de los conductillos intra y extralobulillares, mediante un intercambio activo de HC03- por Cl- (1) (Fig.2).
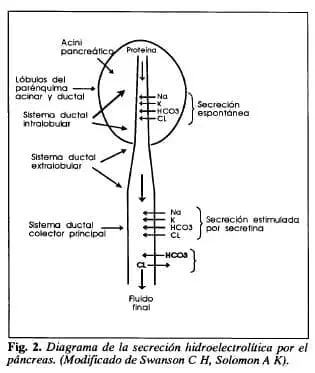
Bajo condiciones de estimulación máxima, el volumen de fluido pancreático y la concentración de bicarbonato se mantienen estables; a menores grados de estimulación se encuentran mayores grados de variación en la concentración de bicarbonato en el fluido pancreático, pues al permanecer un mayor tiempo la secreción en contacto con las células ductales del sistema colector principal, éstas pueden influir de manera más prolongada en la composición de estos elementos. Estas variaciones en la composición de la secreción hidroelectrolítica del páncreas, llevó a hacer suponer y a demostrar posteriormente la existencia en el sistema ductal pancreático, de una barrera de mucopolisacáridos que, al igual que en el estómago, permite la retrodifusión de iones. Esta barrera, como la gástrica, es sensible a la acción de diferentes noxas (aspirina, alcohol, sales biliares) que alteran la capacidad del dueto de mantener altas concentraciones de bicarbonato en el fluido pancreático (11).
Fisiopatologia de la pancreatitis
Hay dos interrgogantes importantes en la fisiopatogenia de la pancreatitis aguda.
El primero hace relación al mecanismo que permite la activación intrapancreática de las enzimas y el acceso de las mismas al intersticio glandular.
El segundo se relaciona con los grupos enzimáticos más importantes en la producción de la necrosis tisular.
En tanto que el segundo interrogante parece haber sido resuelto con alguna claridad, la respuesta al primero aún permanece confusa.
Similarmente hay poca formación relacionada con los mecanismos que influyen en la progresión de las formas edematosas a las más severas, hemorrágica y necrótica.
Bajo condiciones de normalidad, el páncreas es protegido de la activación intrapancreática de sus grupos enzimáticos por varios mecanismos (12).
1. Las enzimas proteolíticas y la fosfolipasa A, son secretadas como zimógenos y proenzimas inactivas.
2. Los zimógenos de las enzimas proteolíticas están separados de las proteínas celulares por medio de membranas de retículo endoplásmico.
3. Tanto en la secreción pancreática, como en el tejidoglandular, existen inhibidores de las enzimas proteolíticas, como el inhibidor pancreático de la tripsina. Otros inhibidores como la Alfa-1 antritripsina y la Alfa-2 macroglobulina, se encuentran presentes en el plasma.
4. El pH alcalino de la secreción pancreática protege la glándula de la acción de sus propias enzimas. En primer lugár, el tripsinógeno se autoactiva a pH bajo (aproximadamente 5.0) y esta tripsina activada, activa por autocatálisis a todos los demás precursores enzimáticos. En segunda instancia, el inhibidor proteico de la tripsina en pH alcalino permanece enlazado de manera más estable y por un tiempo mayor a las pequeñas cantidades de tripsina activa que eventualmente pudieran encontrarse en el fluido pancreático (2).
La tripsina, además de causar la destrucción proteolítica del parénquima pancreático, convierte el kalicreinógeno en kalicreína, produciendo en forma indirecta bradiquinina. Influye sobre los sistemas de coagulación y del complemento mediante la activación del factor de Hageman. Estos factores contribuyen a incrementar la inflamación local con aparición de trombosis capilar, lesión tisular por hipoperfusión e hipoxia, hemorragia y las demás manifestaciones sistémicas de la pancretitis aguda (P A).
La lipasa provoca necrosis grasa en la P A: su papel en el origen y en la cadena de eventos que ocurren en esta entidad no es suficientemente claro; pero hay. alguna evidencia de que adquiere importacia cuando la cascada de alteraciones bioquímicas se ha iniciado.
La salida de elastasa del sistema ductal, determina la destrucción de las fibras elásticas de los vasos sanguíneos favoreciendo la aparición de hemorragia. La fosfolipasa A favorece la destrucción de la membrana celular y al actuar sobre la lecitina de la bilis produce lisolecitina, elemento altamente tóxico para la barrera del sistema ductal del páncreas.
Teorías corrientes sobre la génesis de la pancreatitis
Múltiples teorías se han propuesto para explicar el origen de la pancreatitits desde la del canal común propuesta por Opie, hasta la del reflujo biliar dentro del canal pancreático, pasando por la de la obstrucción e hipersecreción, todas ellas relacionadas con altera iones de la barrera celular, y más recientemente aparece la teoría de la colocalización que llevaría la génesis de la pancreatitis al nivel ultraestructural dentro de los componentes de la célula del acinipancreático (8).
Aisladamente parece ser que ninguna de las teorías relacionadas con la lesión de barrerá, explica completamente la cadena de eventos bioquímicos de la entidad que nos ocupa, sino que hay situaciones diferentes según el factor etiológico desencadenante. De manera resumida se presentarán las diferentes hipótesis existentes con relación al origen de la pancreatitis aguda, según su factor desencadenante.
Enfermedad biliar
La enfermedad del tracto biliar se encuentra asociada a la pancretitis aguda en porcentajes muy variables, que pueden fluctuar entre el 5% (13) Y el 75% (14) de acuerdo con las series que se consulten. En nuestro medio, se encuentran porcentajes de incidencia que oscilan entre el 8% en la serie de García y col (15) y un 52.1 % encontrada en nuestra casuística en el Hospital Militar Central de Bogotá (16, 17).
En 1901 Opie describe dos pacientes que fallecen de pancreatitis aguda, a quienes durante la n~cropsia, se les encontró un cálculo biliar impactado en la ampolla de Vater y propone su teoría del “canal común”, argumentando que el reflujo de bilis a los canales pancreáticos podría haber iniciado el proceso de pan crea titis aguda. Posteriormente, se demostró que a presiones fisiológicas, la presencia de bilis fresca en los canales pancreáticos no inicia la cadena de eventos bioquímicos que caracteriza a la pancreatitis aguda (18).
También se propuso la teoría de que el cálculo impactado en la ampolla permitiría el reflujo de enzimas dentro del árbol biliar; experimentalmente se demostró que la incubación de bilis y enzimas pancreáticas produce una mezcla altamente tóxica posiblemente por la presencia de Iisolecitina (19), que en unión de las sales biliares lesionarían la barrera mucopolisacárida del ducto pancreático, permitiendo el paso de grandes moléculas de enzimas digestivas dentro del parénquima pancreático (11).
A pesar de los conceptos enunciados anteriormente, hay una serie de observaciones que se oponen a la teoría del canal común, como única explicación al origen de la pancreatitis aguda de causa litiásica biliar; de acuerdo con Quinlan entre otros, el canal común sólo se encuentra en una minoría de los pacientes; el 70% de los casos presentan una papila accesoria que marca la desembocadura del canal de Santorini (9, 20); adicionalmente se demostró que la presión secretoria pancreática excede a la presión secretoria biliar (21); este hecho, en presencia de una obstrucción de la ampolla de Va ter, favorecería más el reflujo de la secreción pancreática dentro del árbol biliar que la situación inversa. Como argumento final en contra de la teoría del “canal común” se encuentra el hecho de que la bilis por si sola, no activa las proenzimas secretadas por el páncreas (22).
Acosta (23) Y otros investigadores (24) hablan del alto porcentaje de cálculos biliares recuperados por las heces de pacientes aquejados de P A de etiología biliar y estos hallazgos han permitido proponer que la migración del cálculo interfiere, así sea en forma transitoria, el mecanis mo esfinteriano de la ampolla permitiendo el reflujo de contenido intestinal dentro del confluente biliopancreático (18, 24, 25), desencadenándose una serie de reacciones bioquímicas que se comentan a continuación.
La enteroquinasa contenida en el jugo duodenal activa las formas pro de las enzimas proteolíticas, permitiendo la aparición de tripsina activada, nociva para la barrera mucosa del conducto pancreático. La B-glucuronidasa de los grupos coliformes presentes en el líquido duodenal, actuando sobre las sales biliares, produce sales biliares no conjugadas, tóxicas también para la barrera ductal (26). Finalmente, la fosfolipasa A del grupo lipolítico de la secreción pancreática al actuar sobre la lecitina de la bilis, produce lisolecitina, igualmente tóxica para la barrera mucosa del conducto (19). La concepción artística de esta serie de eventos se muestra en las Figs. 3 y 4.
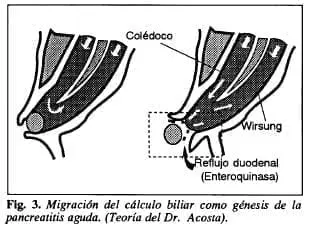
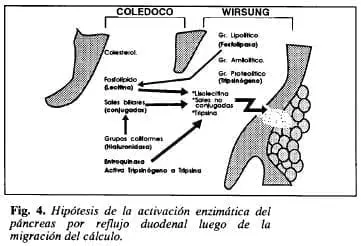
En este punto y teniendo en cuenta los conceptos presentados durante el análisis de la fisiología normal, podría agregarse que la lesión de la barrera ductal por estas diferentes noxas, al alterar la capacidad de secreción de bicarbonato al fluido ductal, con la consiguiente disminución del pH del líquido pancreático, facilitaría la autoactivación del tripsinógeno a tripsina, disminuyendo, además, la estabilidad de los enlaces del inhibidor proteico de la tripsina.
Esta teoría que integra una serie de conceptos planteados por diferentes investigadores, tiene opositores, y el más serio argumento hace relación a la poca frecuencia con que la pancreatitis aguda aparece luego de cirugías realizadas sobre el esÍmter (esfinteroplastias) o a continuación de pancreatoyeyunostomías.
En lo personal y desde un plano eminentemente teórico, consideramos que el solo reflujo duodenal no es el responsable de toda esta cadena de eventos; se requiere la hipertensión intraductal generada por la impactación del cálculo, así sea de manera transitoria, que facilite el reflujo pancreático al árbol biliar, permitiendo la acción de la fosfolipasa sobre la lecitina biliar, para formar lisolecitina, tóxica para la barrera del ducto. El íleo reflejo en el asa duodenal, secundario al proceso i.nflamatorio, permitiría la proliferación de gérmenes coliformes, hecho demostrado por Pfeffer al crear un asa duodenal cerrada que ocasionó la aparición de pancreatitis aguda en el modelo experimental (8). Al migrar el cálculo, el contenido duodenal rico en enteroquinasa y con un aumento en la densidad de la flora coliforme, agregaría los otros dos factores necesarios para desencadenar el proceso de pancreatitis aguda secundaria a patología litiásica biliar.
Alcohólica
La pancreatitis aguda a menudo se presenta después de la ingesta de licor, pero el mecanismo mediante el cual se desencadena el proceso pancreático no es bien conocido. El alcohol determina un incremento en la acidez gástrica y en forma secundaria mediante la acción de la secretina causa un aumento en la secreción pancreática; y por acción directa provoca edema y espasmo del esfínter de Oddi. Esto llevó a proponer la teoría de la obstrucciónhipersecreción en la génesis de la pancreatitis.
Cueto y Tajen (27) demostraron que el alcohol produce pancreatitis a pesar de excluir el asa duodenal de su acción directa y prevenir el espasmo ampular. Sarles y posteriormente Sahel (28, 29) comprobaron que la ingesta prolongada de alcohol, origina una alteración de la secreción pancreática en su calidad, por un incremento en la producción de proteínas con tendencia a la formación de trombos proteicos en el sistema ductaI. Estos trombos se calcifican dando origen a minicálculos en los ductos secundarios y terciarios que van a lesionar el epitelio de los mismos y de los acinis, proceso que cursa en forma asintomática, hasta que se presenta el primer episodio de P A de manera tal que a pesar de ser éste clínicamente agudo la entidad es histológiicamente crónica (30).
Cameron, Zuidema, Capuzzi y Margolis (31, 33) observaron alteraciones en el perfil lipídico de alcohólicos admitidos con pancreatitis y propusieron que la hipertrigliceridemia que origina el alcohol es hidrolizada por la lipoproteinlipasa a ácidos grasos libres, los que serían responsables del desarrollo clínico de la pancreatitis.
Kimura y Saharia (34, 35) reprodujeron la pancreatitis de manera experimental en páncreas aislados de perros, mediante la infusión de ácidos grasos libres; iguales resultados a los observados por los anteriores, son informados por Sanfey (36).
Hiperlipidémica
A pesar de lo mencionado anteriormente, el papel de los triglicéridos séricos en la patogénesis de la pancreatitis aguda de origen alcohólico continúa siendo controvertido, pero si está aceptada la asociación entre hiperlipidemia y P A (37), esta asociación generalmente se presenta de acuerdo con la clasificación de Frederickson en las hiperlipidemias tipos 1, IV Y V (38).
Varias teorías se han propuesto para explicar los mecanismos mediante los cuales los triglicéridos séricos predisponen a la aparición de pancreatitis. Havel y otros (39, 41) propusieron que la lipoproteinlipasa, presente en altas concentraciones en los capilares y linfáticos pancreáticos, puede producir una rápida hidrólisis de los triglicéridos. Esto conduciría a altas concentraciones de ácidos grasos libres, que se precipitarían en forma de microtrombos en los capilares pancreáticos, ocasionando isquemia o ejerciendo efecto tóxico directo sobre las células acinares. Haig (42) demostró experimentalmente en animales, que las dietas con alto contenido de lípidos predisponen a la pancreatitis. Sus estudios demostraron que la susceptibilidad no estaba relacionada con cambios en la calidad o cantidad de la secreción pancreática y sugirió que la integridad de la membrana celular de los acinis podría ser la afectada por la acción de los lípidos.
posiblemente las células acinare.s son más sensibles a ser lesionadas cuando están expuestas a niveles altos de lípidos séricos. En tal situación los triglicéridos séricos elevados o los ácidos grasos libres podrían no iniciar la inflamación pancreática, pero sí predisponer la acción tóxica de algún otro agente.
Medicamentosa
La ocurrencia de P A se ha informado durante la administración de una amplia variedad de drogas. Una relación generalmente aceptada se establece con los corticoides suprarrenales, estrógenos, diuréticos, tiazídicos, azatioprina y furosemida (37).
Nakashima y Mallory (43, 44) han sugerido que la hipertrigliceridemia asociada a la administración de corticoides, estrógenos y tiazidas podrían ser el factor responsable de la pancreatitis.
Posoperatoria
La P A con frecuencia se presenta en forma secudaria a intervenciones quirúrgicas sobre la vía biliar y el segmento gastroduodenal. En estos pacientes es probable que el trauma directo sobre el páncreas, su irrigación o la obstrucción duodenal que facilitaría la proliferación bacteriana, fuesen los responsables del proceso (45, 46). Morgan, Robioson y White (47) describieron disminución en la concentración del inhibidor de la tripsina en el jugo pancreático en el posoperatorio, permitiendo una mayor susceptibilidad del páncreas a la injuria. También se han descrito episodios de pancreatitis aguda luego de derivaciones cardiopulmonares y trasplantes cardíacos; se considera que en estos casos hay una perfusión inadecuada en la microcirculación pancreática durante el tiempo de la derivación, lo que favorece la aparición de pancreatitis secundaria a los fenómenos isquémicos; adicionalmente se cree que la isquemia puede ser un factor contribuyente, pues la lesión por frío es reconocida como causa de pancreatitis (48, 49).
Traumática
El trauma directo sobre el páncreas se presenta con alguna frecuencia en los traumatismos abiertos o cerrados del abdomen. La inflamación pancreática secundaria se encuentra relacionada con la ruptura de los conductos pancreáticos y la isquemia parenquimatosa secundaria al trauma (37).
Hiperparatiroidismo
Tradicionalmente se ha asociado el hiperparatiroidismo con episodios de P A Y tres hipótesis se han presentado para explicar esta relación. La primera plantea la formación de cálculos cálcicos en el interior del ducto pancreático. La segunda hipótesis manifiesta que los niveles elevados de parathormona pueden tener efecto tóxico sobre el páncreas. La tercera postula que los niveles elevados de calcio en la secreción pancreática, pueden acelerar la conversión de tripsinógeno en tripsina (50,51).
Sin embargo, los cálculos pancreáticos no se encuentran de manera uniforme en pacientes afectados de hiperpara tiroidismo y otras situaciones que producen hipercalcemia como el mieloma múltiple y la sobredosificación de calcio parenteral, también pueden llegar a provocar episodios de pancreatitis aguda (52,53).
La validez de esta asociación no es unánimemente aceptada, y Bess en l;studio presentado sobre 1.153 pacientes con hiperparatiroidismo, sólo encontró episodios de pancreatitis aguda en el 1.5% de los casos, y el 65% de este grupo presentó factores predisponentes adicionales a su enfermedad pancreática (54). Watson y col, sobre 100 pacientes sometidos a paratiroidectomía, no encuentran episodios francos de P A aunque informan 4 casos de hiperamilasemia transitoria (55).
Isquemia
La reducción parcial de moléculas de oxígeno en los sistemas biológicos producen intermediarios citotóxicos como los superóxidos (Oz), el peróxido de hidrógeno (HzOz) y los radicales hidroxilo (OH). Tales sutancias juegan importante papel en una serie de estados patológicos incluyendo las lesiones posisquemia, la intoxicación por oxígeno, los estados posradiación y la inflamación mediada por fagocitos (56).
Esta hipótesis planteada respecto a la fisiopatología de la P A se refiere a los fenómenos isquémicos; es atrayente pues logra plantear un hecho común aplicable a cualquier factor desencadenante. Broe, Zuidema y Cameron (57) trabajando con páncreas canino aislado, sometieron algunos a diferentes flujos con presiones controladas de oxígeno y a otros a isquemia total y encontraron diferencias estadísticamente significativas en cuanto a aumento de peso y excreción de amilasa en los totalmente isquémicos, respecto a los demás.
Sanfey y col (58) reprodujeron las experiencias de los anteriores, logrando bloquear los cambios isquémicos al adicionar alopurinol (inhibidor de la xantinoxidasa). Propusieron que la vía de las xantinas sería la productora de radicales libres de oxígeno, que actuarían como mecanismo gatillo ante la hipoxia severa ..
Parks y col (59,60) demostraron el efecto de tales radicales en nivel de circulación capilar, en donde al lesionar el endotelio capilar se determina la aparición de trombosis que excluyen amplios segmentos de la microcirculación. Estas alteraciones han sido demostradas prácticamente en todos los tejidos de la economía por mecanismos fisiopatológicos similares (56,61-63).
Teoría de la colocalización
Todas las teorías anteriormente mencionadas, de una manera u otra, se relacionan con lesión de la barrera Iipídica celular, por diferentes factores y eventos que conducen a la autodigestión pancreática en el espacio extracelular; en un artículo recientemente publicado, Steer (8) propone la activación intracelular de las enzimas pancreáticas por las hidrolasas Iisosómicas, como posible mecanismo fisiopatológico; se sugiere con base en estudios experimentales, que alteraciones en la secreción y transporte de las proteínas enzimáticas jugarían papel importante en la evolución de la pancreatitis aguda.
Biología celular
Las células del páncreas como todas las del organismo humano, son de tipo eucariótico y caracterizadas por su alta compartimentalización. Entre sus muchos compartimientos intracelulares, ninguno es más complejo que el sistema reticuloendoplásmico, constituido por una red de canales o espacios rodeados de membranas que llenan el citoplasma hasta el plasmalema. De otra parte los lisos 0- mas celulares son estructuras sacciformes que contienen en su interior enzimas digestivas, que actúan como el sistema digestivo intracelular. Se han logrado identifiicar en su interior por lo menos 40 ezimas digestivas que incluyen: proteasas, nucleasas, glucosidasas, Iipasas, fosfolipasas, sulfatasas y fosfatasas; la gran mayoría de estas enzimas son hidrolasas ácidas, óptimamente activas a pH 5.0 y con ellas los lisosomas se encuentran en condiciones de digerir la mayor parte de los componentes básicos de células y tejidos.
Las sustancias que tienen acceso a los lisosomas pueden provenir del exterior de la célula por heterofagia o por permeación directa; o del interior de la célula por autofagia o crinofagia. La heterofagia consiste en la captación de materiales extracelulares por invaginación de la membrana celular o endocitosis, que puede realizarse mediante diminutas invaginaciones, pinocitosis, o en grandes bolsas que pueden llegar a englobar microorganismos, fagocitosis. Los componentes celulares pueden tener acceso a los Iisosomas por fusión directa, por ejemplo, de vesículas o gránulos secretores, crinofagia, o por autofagia que consiste en la captación masiva de pequeñas porciones del citoplasma que a menudo contienen órganos celulares reconocibles, que son rodeados por cisternas de membrana lisa derivadas del aparato de Golgi o del reticuloendotelio (64).
Síntesis y secreción de proteínas enzimáticas
En situaciones de normalidad, la síntesis de las enzimas digestivas y de las hidrolasas se realiza en los ribosomas unidos al reticuloendoplásmico rugoso; a partir de éste la proteína migra hacia el complejo de Golgi y las enzimas se almacenan en vacuolas que maduran en gránulos de zimógeno; por su parte las hidrolasas se depositan en el interior de los Iisosomas.
Las enzimas al ser secretadas como precursores inactivos, se encuentran almacenadas en vacuolas que contienen en su interior potentes inhibidores enzimáticos que impiden su activación intracelular; estas vacuolas separan a los precursores enzimáticos del citoplasma celular.
Basado en los estudios de Lombardi y Lampel, Steer y su grupo (8), observaron en modelos experimentales sujetos a dietas deficientes en colina o sometidos a estímulos supramaximales con secretagogos, que la síntesis y transporte de enzimas a los gránulos de zimógeno no se alteraban, pero sí su secreción a partir de la célula acinar. Esta situación generaba un aumento en la concentración intracelular de gránulos de zimógeno, que con el tiempo se unían a los Iisosomas mediante un proceso de crinofagia, formándose grandes vacuolas que contenían simultáneamente en su interior, precursores enzimáticos inactivos e hidrolasas Iisosómicas, dentro de las cuales la Catepsina B, es capaz de activar el tripsinógeno.
Iguales observaciones se obtuvieron cuando se bloqueó la secreción pancreática en conejos, mediante la obstrucción selectiva del ducto pancreático. No se apreció modificación en la rata de síntesis enzimática ni en el transporte de las proteínas enzimáticas a los gránulos de zimógeno, pero sí la aparición de grandes complejos vacuoliformes que contenían en su interior, tanto proenzimas inactivas como hidrolasas Iisosómicas (65,66).
ocurre, como se había especulado hasta ahora, en el espacio extracelular, sino en el interior de las células del acini pancreático. Extrapolando esta observación, se podría suponer que la obstrucción ductal secudaria a la impactación de un cálculo o al edema papilar ocasionado por la ingesta de alcohol, pueden generar un bloqueo de la secreción pancreática que de manera retrógrada, provocan hipertensión en todo el sistema ductal del páncreas hasta generar un bloqueo a la secreción de enzimas en nivel de la membrana de las células acinares, facilitando la localización de las hidrolasas Iisosómicas, simplificando de esta manera la activación intracelular de las enzimas digestivas (67). Esta hipótesis, junto con la teoría de los radicales libres de oxígeno, cambia por completo todo el enfoque actual en el manejo de la progresión de la pancreatitis aguda, pues la autodigestión pancreática no ocurre, como se había especulado hasta ahora, en el espacio extracelular, sino en el interior de las células del acini pancreático.
Abstract
Current concepts on the Physiopathology of acUle pancreatitis are reviewed. Migration of calculi as the etiologic factor of gallstone pancreatitis is presented.
Ischemia and the production of oxygen free radicals are importan! factors in the evolution of this disease.
Finally the activation of hydrolyses at the lysosomal level is where the process of acute pancreatitis is initiated, this new concept is reviewed in detail.
Referencias
1. Brooks J R: Historical development and epidemiology. In: Brooks J R (00), Surgery of the pancreas. Filadelfia, W. B.
Saunders Company, 1983. p. 25
2. Peterson L M: Biochemistry and physiology of the pancreas. In: Brooks J. R (00)., Surgery of the pancreas. Philadelphia, W. B. Saunders Company, 1983. p. 25
3. Heitz P U, Beglinger C, Gyr K: Anatomy and physiology of exocrine pancreas. In: KJoppel G., Heitz P (eds)., Pancreatic Pathlogy. Londres, Edinburgh, Churchill Livingstone, 1984. pp. 3- 21
4. Case R M: The cellular basis of pancreatic secretory processes. In: Duthrie H L., Wormsley KG (eds)., Scientific basis of gastroenterology. Edimburgh, Churchill Livingstone, 1977
5. Cubilla A L, Fitzgerald P J: Morphological patems of primary nonendocrine human pancreas. Cancer Res 1975; 35: 2234-48
6. Desnuelle P, Figarella C: Biochemistry. In: Howard HT, Sarles H (eds). The exocrine pancreas, London, Saunders, 1979
7. Brooks F P: Applied physiology of the pancreas exocrine. In: Diseases of the pancreas. Practical Gastroenterology. Long Island City. N.Y. 1978, pp. 4-6
8. Steer M L: Clasification and pathogenesis of pancreatitis. Surg Clin North Am 1989; 69: 467-80
9. Pincus 1 J:’ Embriology, anatomy and phisiology of the pancreas. In: Bockus H L (ed), Gastroenterology, 2a Ed. Philadelphia, Saunders, 1966
10. Sleisenger M H: Fisiopatología del tracto gastrointestinal. En: Smith L H, Thier S O (eds). Fisiopatología. Principios biológicos de la enfermedad. Buenos Aires. Editorial Médica Panamericana 1983: 1282-91
11. Reber H A, Roberts C, Way L W: The pancreatic duct mucosal barrier. Am J Surg 1979; 137: 128-32
12. Gyr H, Heitz P U, Beglinger C: Pancreatitis. In: KJoppel H, Heitz P U (oos): Pancreatic Pathology. London, Churchill Livingnstone, 1984 pp 44-72
13. Howes R, Zuidema G O, Cameron J L: EvaIuation of prophylactic antibiotics in acute pancreatitis. J Surg Res. 1979; 18: 197-200
14. Banks P A: Pancreatitis. Trib Méd 1982; 64:29-38
15. García F, Méndez C G, Arteta O S: Pancreatitis aguda en el Hospital Universitario de Cartagena. Estudio de 224 casos. 1976-1989. Rev Col Gastroent 1991; 6 (3): 163-71
16. Reyes R., Gímez M: Pancreatitis aguda. Revisión de 48 casos, de 1968 a 1973. Temas Escogidos de Gastroenter~logía 1975; 18:185-95
17. Nieto J A., Castelblanco J, Pimiento H: Pancreatitis aguda. Casuística del Hospital Militar Central de Bogotá. Rev Col Cirug 1989; 4 (3):101-10
18. McCutcheon A O: A fresh approach to the patogenesis of pancreatilis. Gut 1968; 9:296-300
19. Poncelet P, Thompson A G: Action of bile phospholipids on the pancreas. Am J Surg 1972; 123: 196-9
20. Quinian R M: Anatomy, embriology and physiology of the pancreas. In: Shackelford R T, Zuidema G O (eds): Surgery of the alimentary trac!. 2a Ed. Philadelphia, W. B. Saunders 1983: pp. 3-24
21. Menguy R B, Hallenbeck G A, Bollman J L et al: Intraductal pressures and sphinteric resistance in canine pancreatic and biliary ducts after various stimuli. Surg Gynecol Obstet 1958; 106: 306-20
22. Robinson T M, Dunphy J E: Continuous perfussion of bile protease activators through the pancreas. JAMA 1963; 183: 530-3
23. Acosta J M, Pellegrini L A, Skiner O B: Etiology and pathogenesis of acule biliary pancreatitis. Surgery 1980; 88: 118-25
24. Taylor T V, Rimmer S: Pancreatic duct reflux in patienl with gallstone pancreatitis? Lancet 1980: 848-50
25. Najarian J, Delaney J (eds): Cirugía del hígado, páncreas y vías biliares. Barcelona, Editorial Científico Médica 1978; 243-7
26. Williams L F jr, Byme J J: The role of bacteria in hemorragic pancreatitis. Surgery 1968; 64: 967-72
27. Cueto J, Tajen N, Zimmerman B: Studies of experimental alchoholic pancreatitis in the dogo Surgery 1964; 62: 159-62
28. Sarles H: Chronic calcifying pancreatitis-Chronic alcoholic pancreatitis. Gastroenterology 1974; 66: 604-16
29. Sarles H, Sahel J: Pathology of chronic calcifying pancreatitis. Am J Gastroentero11976; 66: 117-39
30. Banks P A: Pancreatitis. Trib Méd 1982; 64:29-38
Doctor Julio Alberto Nieto Silva, Prof. Tit. de Cirugía, Esc. Mil. de Medicina, Hosp. Militar Central, Bogotá, D.C., Colombia.